Chapter 7 Cytokines and Interferons in Lupus
Properties of Cytokines and Their Receptors
Cytokines are small soluble proteins that are produced by immune system cells and mediate activation or functional regulation of nearby cells by binding to cell surface receptors.1 In health, the immune system functions as a coordinated whole, with each cell type playing a carefully orchestrated role. These molecules mediate the communication between immune cells, which is critical for coordinated responses to pathogens. Cytokines are important at each stage of the immune response, from the initial activation of the innate immune system, through the maturation of T and B cells in the adaptive response, to the resolution of the immune response once the pathogen is cleared. Given these important roles, it is easy to imagine that inappropriate cytokine signaling could lead to autoimmunity. Both excessive inflammatory cytokine production and insufficient inhibitory cytokine production likely play a role in the chronic autoimmune inflammation that characterizes SLE. Abnormal cytokine signaling may participate in the initiation of SLE, and cytokines are also important in the progression and propagation of the illness.
Cytokines act in an autocrine or paracrine manner, in close proximity to their source, and have been considered to serve a similar function to that of neurotransmitters in the nervous system. Basal production of most cytokines is negligible, and activation of the producer cells, through pattern-recognition or antigen-specific receptors, results in either new gene transcription or translation of preexisting cytokine messenger RNAs (mRNA) and protein secretion. Many cytokines have been reported to be elevated or decreased in the sera of patients with SLE in comparison with healthy controls (Table 7-1). Cytokine protein is typically detected at low levels or not at all in serum from healthy individuals, but in patients with SLE, elevations of some cytokines are measurable and some vary with disease activity. These findings should reflect the immune dysregulation that characterizes the disease to some degree. Presumably the cytokines observed in the circulation come from the inflamed tissues, although some cytokines could be produced in the blood as well.
TABLE 7-1 Role of Cytokines and Interferons in Lupus Pathogenesis
| MEDIATOR | ROLE IN PATHOGENESIS |
|---|---|
| Products of the Innate Immune Response | |
| Type I interferon: IFN-α, IFN-β, IFN-ω | |
| Tumor necrosis factor | |
| Interleukin-1 (IL-1) | Increased levels associated with active SLE |
| IL-10 | |
| BLyS/BAFF (B-lymphocyte stimulator/B-cell–activating factor) | |
| IL-6 | |
| IL-12 and IL-18 | Support expansion of T helper 1 (Th1) cells and natural killer (NK) cells |
| IL-8, IP-10, MIG (monokine induced by IFN-γ), MCP-1 (monocyte chemotactic protein 1), fractalkine | Chemokines that may be increased in active SLE and may recruit inflammatory cells to sites of organ inflammation, particularly in lupus nephritis |
| Products of the Adaptive Immune Response | |
| IL-2 | |
| IFN-γ | |
| IL-6 and IL-10 | Produced by T and B lymphocytes as well as monocytes |
| TGF-β (tumor growth factor beta) | |
| IL-17 | |
| IL-21 |
The level of cell surface expression of the receptors to which cytokines bind is also highly regulated and contributes to the impact of the cytokines on immune system activity. Once the receptors are engaged, complex multicomponent molecular pathways transduce a signal from cell surface to nucleus, resulting in new gene transcription. The Janus kinase (Jak)–signal transducer and activator of transcription (STAT) pathways are common mediators of cytokine-cytokine receptor interactions.2 The strength of these signaling pathways can be affected by the state of activation of the target cell and the additional signals that it has received, with the mitogen-activated protein (MAP) kinase and other signaling systems modulating the function of the Jak-STAT pathway. In addition, negative regulators of cytokine signals, such as the suppressor of cytokine signaling (SOCS) proteins, further modulate the strength of target cell response to the cytokine.3
The degree of expression of individual cytokines or activation of their pathways is also regulated on that basis of genetic differences among individuals that translate into variable efficiency in cytokine production or response.4 Great progress has been made in understanding the genetic basis of SLE, and many of the well-established genetic risk loci are genes that function in cytokine pathways.5,6 Genetic variability can be localized to regulatory regions of genes, potentially modifying the level of expression, or can be in coding sequences, sometimes resulting in an altered amino acid sequence and modified conformational structure. Both of these types of genetic variations are represented in the list of genetic associations with SLE, and some of these SLE-associated variations are also associated with altered cytokine levels, supporting the idea that genetic variability in cytokine response is a component of SLE pathogenesis.6
Assessment of Cytokine Production
The expression of cytokines and the capacity to produce cytokines in an individual can be assessed using numerous distinct and complementary approaches.7 Measurement of an mRNA encoding a cytokine can be used to provide a reasonable indication of the amount of cytokine protein produced. However, the variable stability of one or another mRNA must be considered, and the presence of an mRNA may not necessarily indicate that the mRNA is translated and the corresponding protein generated.
Use of Microarray to Study Cytokine Effects
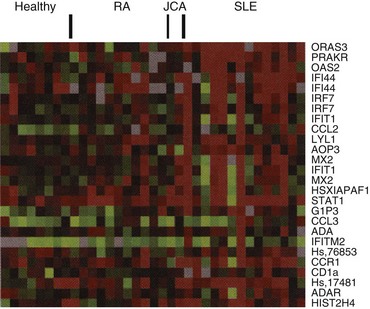
Microarray analysis, a system in which thousands of oligonucleotide sequences are spotted on a solid substrate, usually a glass slide, and RNA-derived material from a cell population is hybridized to the gene array, is an innovative technology that permits a global view of the profile of genes expressed in a cell population at a point in time, including genes in the cytokine pathways.8 Applying microarray analysis to heterogeneous cell populations in peripheral blood from patients with autoimmune diseases raises technical challenges. The variable proportions of different cell populations in each subject, each making variable contributions to the mRNAs in the blood sample, adds complexity to the comparison of study groups. Additionally, the statistical analysis of thousands of gene sequences studied in multiple individuals is daunting. Investigations have now demonstrated that in spite of these technical challenges, significant and useful microarray data can be derived from complex cell samples, including peripheral blood mononuclear cells stimulated in vitro and peripheral blood preparations from patients with autoimmune disease. The view that IFN-α might play a central pathogenic role in SLE has only lately gained momentum with the completion of several large-scale studies of gene expression profiling with microarray technology. Multiple groups have used this powerful technology to demonstrate that mRNAs encoded by IFN-regulated genes are among the most prominent observed in peripheral blood cells of patients with lupus (Figure 7-1).8–12 This coordinate overexpression of multiple IFN-α–induced transcripts is what would be expected following ligation of the type I IFN receptor by IFN-α, and this molecular footprint has been called an “IFN-α signature.”
Activation of the Immune Response in SLE
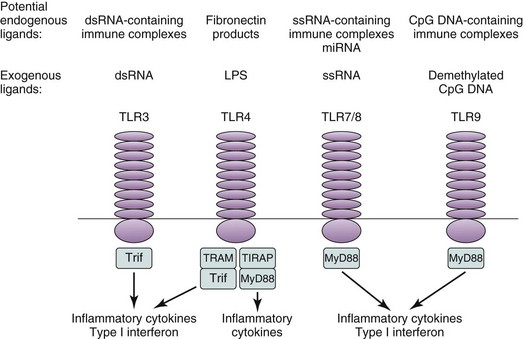
The mechanisms that account for aberrant production of autoantibodies, cytokines, and other soluble mediators in SLE can be modeled in parallel with the production of antibodies and mediators in a productive immune response to microbial pathogens in a healthy individual. The initial encounter with the microbe is mediated by cells of the innate immune response. Although those cells have traditionally been considered to initiate an immune response through nonspecific cell surface receptors, the elucidation of the Toll-like receptor (TLR) family has altered that picture.13 Although the innate immune response does not have the fine level of specificity that characterizes the adaptive immune response generated by T and B lymphocytes, the members of the TLR family do recognize classes of stimuli with characteristic structural features. Among the TLR ligands are nucleic acids, including hypomethylated CpG DNA and single-stranded (ss) or double-stranded (ds) RNA. These nucleic acids are typical components of viruses and bacteria but might also be products of apoptotic or necrotic host cells. Whether microbe-derived in the setting of infection or self-derived, these oligonucleotides provide an adjuvant-like stimulus that can initiate a heightened level of immune system activity, including the production of cytokines.
Cytokines of the Innate Immune Response
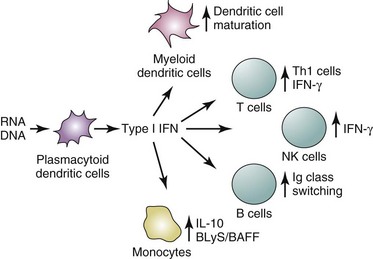
Later studies support a contribution of signals through TLRs to the activation of the innate immune response in lupus.14–19 Among the documented triggers relevant to SLE are immune complexes containing DNA or RNA, along with specific antibodies.16–19 A consequence of TLR ligation is production of type I IFN, predominantly IFN-α, which then mediates numerous functional effects on immune system cells. Plasmacytoid dendritic cells (PDCs), a rare cell type that is enriched in skin lesions of lupus patients, are presumed to be active producers of IFN-α.20–24 Interaction of IFN with widely expressed cell surface receptors activates intracellular signaling pathways and induction of transcription of a large number of IFN-responsive genes, including those associated with maturation of myeloid dendritic cells. The result is predicted to be increased APC function and augmented capacity to trigger self-reactive T cells.25
In addition to plasmacytoid and myeloid dendritic cells, mononuclear phagocytes are essential for the inactivation of pathogenic infectious organisms and for the clearance of potentially pathogenic immune complexes and senescent or apoptotic cells. These cells are also important in SLE. Impaired clearance of apoptotic cells has been demonstrated in some studies of patients with SLE, and IFN-mediated maturation of monocytes into effective APCs has been shown in another study.25,26 Macrophages bind, process, and present antigenic peptides to T cells; they physically interact with T cells, delivering secondary activation signals through cell surface adhesion and co-stimulatory molecules; and they secrete a panoply of soluble products, including tumor necrosis factor (TNF), interleukin-1 (IL-1), IL-6, IL-10, IL-12, and B-lymphocyte stimulator (BLyS), that provide important accessory and regulatory signals to both T and B cells. The roles of these products of innate immune system cells are highlighted here, with a particular emphasis on the type I IFNs.
Type I Interferons
Productive infection of host cells by a virus, leading to synthesis of RNA or DNA molecules of viral origin, induces production of host proteins, including the IFNs. The function of these proteins is to inhibit viral replication and to modulate the immune response to the virus, with the aim of controlling infection. The type I IFN locus on chromosome 9p21 comprises genes encoding 13 IFN-α subtypes as well as IFN-β, IFN-ω, IFN-κ, and IFN-ε, the last mostly restricted to trophoblast cells and produced early in pregnancy.27,28 The IFN-α gene complex is likely to have been generated by repeated gene duplications and recombinations. Although the need for and function of each of the IFN-α genes are not clear, specific virus infections are associated with induction of one or another IFN-α.29,30 Data have now identified additional IFNs that are encoded by a gene family related to the classic type I IFNs.31,32 IFN-λs (IL-28 and IL-29) have only moderate sequence similarity to IFN-α, bind to a distinct receptor, yet induce genes similar to those induced by IFN-α. The relative functional roles of IFN-λ and the chromosome 9p–encoded IFNs are under study.33
IFNα can probably be produced by all leukocytes, but PDCs are the most active producers. Rapid progress in study of type I IFN regulation indicates that cell type (PDC vs. fibroblast), stimulus (dsRNA, ssRNA, DNA), and signaling pathway used (TLR3 vs. TLR7/8 vs. TLR9) all contribute to determining the specific IFN isoforms that are produced.33–45 The TLR family of innate immune system receptors and their downstream signaling components play a central role in mediating activation of type I IFN gene transcription (Figure 7-2). TLRs 7, 8, and 9 signal through the MyD88 adaptor. IFN regulatory factors and additional transcription factors, including nuclear factor kappa B (NF-κB) and activating transcription factor 2 (ATF2), bind to and activate an IFN-stimulated response element (ISRE) present in the IFN-α and IFN-β gene promoters.35–37 Tracking the specific intracellular factors that mediate transcription of specific IFN isoforms may provide clues to the innate immune system receptors and the relevant triggers that drive production of those IFNs.
Type I IFN production represents the first line of defense in response to viral infection. Following invasion of the host by a virus, IFN-α is secreted by PDCs, along with other immune system cells, and binds its receptor on many target cells, resulting in engagement of intracellular signaling molecules and induction of a gene transcription program.46 The IFNs were used as model cytokines when Darnell’s group defined the requirements for cytokine-mediated signal transduction.47–49 Binding of IFN-α to its cell surface receptor was shown to activate Jak1 and then STAT1. Subsequently, it was shown that Tyk2, also a Jak kinase, is constitutively associated with the α subunit of the type I IFN receptor (IFNAR), whereas Jak1 is associated with the β subunit of the receptor. Cytokine binding leads to activation of Tyk2 and Jak1 and phosphorylation of the α receptor subunit and part of the β subunit. Subsequent events include activation of STATs 1, 2, and 3, the insulin receptor substrate proteins 1 and 2 (IRS1 and IRS2), and vav (a protein in guanine nucleotide exchange factor of cell signaling).50 STAT1-to-STAT1 and STAT1-to-STAT2 dimers bind to the pIRE element and ISGF3 (interferon-stimulated gene factor 3), including STAT1and STAT2, and a third protein, p48, binds the IFN-stimulated response element.35,48 The Jak-STAT pathway seems to be sufficient to mediate the antiviral effect of IFN-α, whereas the IRS proteins, as well as other undefined factors, are also required for the antiproliferative effect of IFN-α.50
Activation of the type I IFN pathway has diverse and numerous functional effects on immune system cells. IFN-α matures DCs by inducing expression of intercellular adhesion molecule 1 (ICAM-1), CD86, MHC class I molecules, and IL-12p70, and promotes expression of some T-cell activation molecules.51–53 However, IFN-α has antiproliferative effects on T cells, and it is generally described as a suppressor of T-cell immune activity. IFN-α inhibits expression of some proinflammatory cytokines, including IL-8, IL-1, and granulocyte-monocyte colony-stimulating factor (GM-CSF), and it preferentially promotes T helper 1 cell responses by decreasing IL-4 and increasing IFN-γ secretion.54,55 In the setting of coculture of monocytes with lipopolysaccharide (LPS) or CD4+ T cells with anti-CD3 and anti-CD28 monoclonal antibodies, IFN-α augments IL-10 production.54,56 IFN-γ does not have these effects and in fact inhibits IL-10 production. Although IL-10 has important suppressive effects on T-cell proliferative responses, its capacity to promote B-cell proliferation and immunoglobulin class switching suggests that IFN-α may favor antibody production.57–63 Finally, IFN-α leads to increased natural killer (NK) and T-cell–mediated cytotoxicity.64–66 This effect on cytotoxic T lymphocyte (CTL) function has been exploited in the treatment of several malignancies with IFN-α in order to augment tumor lysis, although the mechanism that accounts for the increased killing has not been elucidated fully. At least one such mechanism is the induction of FasL expression on natural killer (NK) cells and increased Fas-mediated apoptosis.66 IFN-α can also promote an inflammatory response. Among IFN-α–inducible gene targets are several chemokines, soluble mediators that attract lymphocytes and inflammatory cells to tissues. In brief summary, IFN-α helps initiate an adaptive immune response that results in increases in cytotoxic T- and NK-cell activity, Fas-mediated apoptosis, and antibody production and inflammation but decreased T-cell proliferation. Many of these immune system effects are reminiscent of those observed in patients with SLE (Figure 7-3).
Several sets of compelling data suggest an important pathogenic role for IFNs in SLE. Papers published as early as 1979 described increased serum levels of IFN in patients with SLE, particularly those with active disease.67–71 At that time, the distinct type I and type II IFNs had not yet been documented, but within several years IFN-α was cloned, and it became clear that IFN-α was present in particularly high levels in SLE blood. Soon after, it was observed that tubuloreticular-like structures in the renal endothelial cells of patients with SLE and in murine lupus models were associated with IFN-α and that in vitro culture of cell line cells with IFN-α-induced similar intracellular structures.72 These observations suggested not only that IFN-α was increased in concentration in SLE blood but also that it might have a functional impact on cells and perhaps contribute to disease. Another key observation was first reported in 1990 and has been noted many times subsequently. Therapeutic administration of IFN-α to patients with viral infection or malignancy occasionally results in induction of typical lupus autoantibodies and, in some cases, clinical lupus.73–77 This demonstration of induction by IFN-α of SLE in some individuals indicated that given the appropriate genetic background and perhaps in the setting of concurrent stimuli, SLE could be induced by IFN-α. Twenty percent to 80% of patients treated with IFN-α have been noted to demonstrate autoantibodies specific for thyroid or nuclear antigens, including anti-DNA autoantibodies.78 Clinically apparent disorders include autoimmune thyroiditis, inflammatory arthritis, and SLE. Hints regarding possible mechanisms of these IFN-α toxicities come from an animal model of autoimmune diabetes.79 Expression of IFN-α by pancreatic islets correlates with development of type I diabetes, and transgenic mice overexpressing IFN-α acquire diabetes. These mice develop autoreactive CD4+ T cells that are Th1 and can kill islet cells. Blanco showed that IFN-α is one component in lupus serum that can promote maturation of blood monocytes to have increased antigen-presenting activity.25 These data are consistent with the demonstration that IFN-α is one of several maturation factors for immature DCs, permitting efficient antigen-presenting function to T cells.80,81 Generation by IFN-α of an APC functional phenotype competent for activation of autoantigen-specific T cells could be an important immune mechanism that incorporates many of these findings.82 Murine studies have supported a role for type I IFN in SLE. Both NZB lupus mice and B6/lpr mice deficient in the IFN-α/β receptor show significant improvement in some manifestations of autoimmunity as well as improvement in clinical disease, and type I IFN promotes development of glomerular crescents and accelerated disease in lupus mice.83–86
In the early 1990s, the major cellular source of IFN-α had not yet been identified, but Ronnblom’s group were able to demonstrate that immune complexes containing lupus autoantibodies and cellular material could induce production of IFN-α by peripheral blood mononuclear cells in vitro.14,20,21,87,88 With the assignment of PDCs as the major source of IFN-α, lupus immune complexes were shown to be active inducers of IFN-α by those cells, and additional data implicate TLR7, TLR9, and the receptor FcγRIIa in the induction of IFN-α by some of those complexes.18–20 Immune complexes containing RNA are particularly effective inducers of type I IFN, presumably through TLR7. Additionally, later data have implicated DNA-containing material derived from neutrophils, referred to as neutrophil extracellular traps (NETs), as stimuli for production of IFN by PDCs.89,90 TLR9 is likely to be the receptor that interacts with those complexes and triggers new gene transcription. A similar scenario has been demonstrated, in a collaboration between two laboratories, in another system relevant to rheumatic diseases, the activation of rheumatoid factor–producing B cells by DNA enriched in CpG immunostimulatory sequences opsonized with anti-DNA antibody.15,16 PDCs appear to be somewhat reduced in the blood, but they have been demonstrated in the skin lesions of patients with lupus.24 It is possible, or likely, that the IFN-α–producing cells have also been recruited to other sites of active disease, including lymph nodes and kidney. Monoclonal antibody to BDCA-2, a cell surface C-type lectin that may contribute to internalization of immune complexes, has been used to identify PDCs.91
Several previous reports documented increased expression of IFN-α–induced genes in SLE, including dsRNA-dependent protein kinase (PRKR) and oligoadenylate synthase (OAS), as well as the protein Mx1, present in lupus-involved skin,92–94 and microarray studies have reproducibly demonstrated that in SLE, IFN-induced genes are the most significantly overexpressed of all those assayed on the microarray.8–12 The type I IFN-inducible gene transcripts are coordinately expressed in lupus peripheral blood, providing strong support for one or more type I IFNs, or a virus-like trigger, as upstream inducers of this gene expression pattern.95 High expression of IFN-inducible genes is seen in approximately 40% to 60% of adult patients with SLE and in a higher proportion of pediatric patients with lupus.10 Adult patients with the IFN signature are characterized by autoantibodies to RNA-binding proteins (Ro, La, Sm, and RNP), higher disease activity, and frequent renal involvement.96 Additionally, younger patients with SLE have higher serum levels of IFN-α,97 and patients of African-American and Hispanic-American ancestry have higher serum IFN-α levels on average than European-ancestry patients with SLE.98 Expression of IFN-α or IFN-inducible gene transcripts or proteins in involved tissue supports the hypothesis that IFN-α contributes to disease pathogenesis in lupus. Of particular interest is the strong IFN gene expression signature observed in SLE synovial tissue in comparison with the pattern seen in rheumatoid arthritis tissue.99
A number of the genetic loci associated with risk of SLE are in or near genes that function in the type I IFN pathway, including IRF5, IRF7, and STAT4,5,100 suggesting that genetic variability among individuals in production and signaling of IFN underlie SLE susceptibility. In fact, a heritable tendency toward high circulating levels of IFN-α has been demonstrated in families with SLE, supporting the idea that genetically determined differences in type I IFN production predispose to SLE.101 Some of the well-established SLE risk genes have been shown to correlate with either higher circulating levels of IFN-α or with increased sensitivity to IFN-α in patients with SLE. For example, in those patients who have autoantibodies that can form immune complexes that trigger the TLR system, the SLE-associated polymorphisms of IRF5102 and IRF7103 result in higher circulating IFN-α levels. These data support the concept that these polymorphisms are gain-of-function in humans, resulting in greater output of IFN-α from the endosomal TLR pathway when this pathway is chronically stimulated by endogenous immune complexes. Other SLE-associated polymorphisms have been associated with a greater sensitivity to IFN signaling in patients with SLE,104,105 resulting in a greater amount of IFN-induced gene expression for a given amount of IFN-α signaling. It is likely that combinations of these relatively common SLE-associated polymorphisms that increase IFN-α production or enhance cellular sensitivity to IFN-α would act in concert in many patients, contributing to the marked IFN pathway dysregulation observed in many patients with SLE. Rare genetic variations resulting in familial lupus may also result in IFN pathway dysregulation. Rare variants in the TREX1 gene have been associated with familial chilblain lupus and SLE.106–108 TREX1 is a nuclease, and the rare variations that are associated with disease are loss-of-function and are thought to result in decreased clearance of nucleic acid. This reduction could lead to activation of the type I IFN pathway via the cytoplasmic nucleic acid receptors or the TLR system, and in fact, the TREX1 deficiency syndromes are characterized by high IFN.109 Given all of the described observations, there is strong support for the hypothesis that inhibition of the type I IFN pathway may benefit patients with lupus, particularly those with increased expression of IFN-inducible genes. However, IFN pathway blockade might weaken the innate and adaptive immune responses to viral infection. Potential approaches to inhibition of the type I IFN pathway could include antibodies specific for the IFN-α receptor or for one or more of the various IFN subtypes noted previously. Other approaches are inhibition of upstream (e.g., TLR pathways) or downstream (e.g., Jaks or STATs) signaling molecules.110 Clinical trials of monoclonal antibodies to IFN-α are currently under way, and initial reports from early-phase studies show inhibition of the IFN signature in skin and blood and some effect on disease activity.111,112

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


