Congenital Scoliosis
Suken A. Shah
Kit Song
INTRODUCTION
Congenital scoliosis is defined as a lateral deviation of the spine associated with one of a broad range of congenital vertebral malformations (CVMs) that can form during in utero development. It is distinct from other spinal deviations in which malformations do not occur and can present as an isolated spine anomaly or be associated with a large number of visceral organ and syndromic abnormalities. In Smith’s “Recognizable Patterns of Human Malformation,” over 40 syndromes have CVMs listed as one of the presenting features (1).
The malformations are always present at birth, but the development of the scoliosis may occur over time. They can occur in any part of the spinal column and are believed to be a result of the disruption of the process of somatogenesis that occurs between the 5th and 8th weeks of gestation (2, 3, 4, 5, 6, 7 and 8). These malformations are not unique to humans as several other species develop such abnormalities in response to teratogenic exposures or from genetic predispositions (9, 10, 11, 12, 13 and 14).
Of great importance is the high incidence of associated abnormalities in other organ systems that can lead to significant adverse impacts upon the health and well-being of affected individuals. In addition to this is the realization that management of the developing deformity is often extremely challenging and, due to the variation of presentation, highly individualized. This chapter focuses upon children with congenital vertebral anomalies. We do not discuss infantile scoliosis or other conditions where there may be deformity without malformations.
INCIDENCE/PREVALENCE
The true incidence and prevalence of congenial vertebral malformations is unknown. Many individuals without visible deformity are undoubtedly not found, and many series intermix patients with syndromic or other neuromuscular conditions. Large population studies utilizing screening chest x-rays for tuberculosis suggested a thoracic spine incidence of 0.5 to 1:1,000 live births (15), with more recent ultrasound screening studies finding an incidence of 0.1 to 0.3:1,000 live births with many of these children having other complex birth defects (16, 17). In general, the incidence should be considered to be rare, with kyphotic CVMs even rarer.
MECHANISM OF SPINE FORMATION
It is useful to review the evolving knowledge and understanding of spine formation and development as it may relate to the formation of CVMs. Much of this knowledge has been gained from the observation of human embryonic development (18) and study of murine models of spine formation. During gastrulation, four identified Hox gene clusters containing 39 genes (Hox A, B, C, and D) are believed to determine positional information along the rostrocaudal axis of developing vertebrates (11, 18, 19). They are expressed in cells of the developing mesoderm and ectoderm that later form the somites, which in turn form the vertebrae, ribs, and muscles. Vertebrae are derived from the paraxial mesoderm that forms from the superficial epiblast cells growing into the primitive streak during gastrulation forming the paraxial mesoderm. These develop into the segmental units of the somite, which then subdivide into ventral sclerotome (vertebral precursor) and dorsal dermatomyotome (muscle, skin, rib precursors) units (3, 18, 20). Induction of the sclerotome is signaled by
the notochord and the floor plate of the neural tube. This involves the Sonic Hedgehog protein (21). Subsequent fusion of the ventral and dorsal sclerotomes then forms vertebrae (3, 18, 20). It is believed that a molecular segmentation clock that is mediated through the Notch signaling pathways controls vertebrate segmentation (3, 10, 12, 22) with formation of somites in a rostral to caudal sequence. This involves periodic expression of 50 to 100 cyclic genes. Genes that are believed to impact this pathway are lunatic fringe (LFNG), DLL3, and MESP2 (23, 24) (Fig. 18-1).
the notochord and the floor plate of the neural tube. This involves the Sonic Hedgehog protein (21). Subsequent fusion of the ventral and dorsal sclerotomes then forms vertebrae (3, 18, 20). It is believed that a molecular segmentation clock that is mediated through the Notch signaling pathways controls vertebrate segmentation (3, 10, 12, 22) with formation of somites in a rostral to caudal sequence. This involves periodic expression of 50 to 100 cyclic genes. Genes that are believed to impact this pathway are lunatic fringe (LFNG), DLL3, and MESP2 (23, 24) (Fig. 18-1).
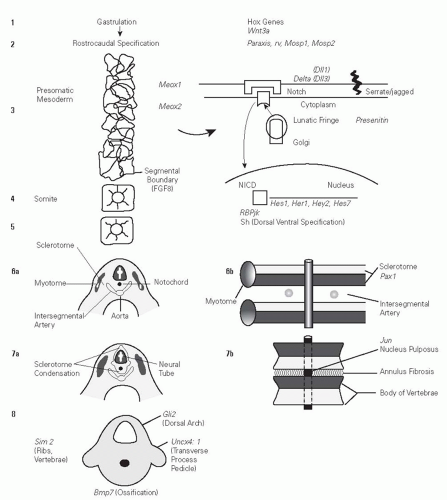 FIGURE 18-1. Schematic diagram showing pathway of murine embryologic development of somites with potential gene interactions highlighted. Segmentation is operated through a molecular clock operating through the Notch signaling pathway. Segment boundary formation is mediated by fibroblast growth factor 8. Sclerotome formation and subdivision into dorsal and ventral halves is mediated by Pax 1 and Meox1. |
ETIOLOGY
The true etiology of the development of CVMs is unknown. The spectrum of the disorder is very large, ranging from syndromic cases such as Jarcho-Levin syndrome to isolated hemivertebrae, and a number of environmental and genetic associations have been identified, suggesting that both genetic factors and teratogenic effects from the environment play a role in the disturbances of normal spine formation.
Anoxia created by carbon monoxide exposure to mouse and chick models at a critical time and specific dose has been demonstrated to cause CVMs very similar to those found in humans (9, 25, 26 and 27). Similar outcomes in humans due to exposure to carbon monoxide have been postulated, but not clearly established (28). Other environmental factors have been clinically associated with the development of CVM. Alcohol exposure with fetal alcohol syndrome (29), anticonvulsant medications Valproic acid and Dilantin (30, 31, 32 and 33), retinoic acid (31), hyperthermia (34), maternal insulin-dependent diabetes (35, 36), and folate deficiency (37) have all been implicated in abnormalities of the spine in humans.
Model organisms such as the chick and mouse strongly suggest that there is a genetic contribution to the development of CVM (3, 11, 12, 38). In humans, the heterogeneous clinical manifestations, variety of morphologic features, and phenotypic presentations described as failures of formation or failures of segmentation had led to the belief that the majority of cases were sporadic events (39). Recent evidence suggests that there is a strong role for genetic factors in the development of human CVMs. The genetic transmission of some cases of CVM has been shown to have a familial recurrence rate of 3% (20, 40), and an increasing number of vertebral malformation syndromes have identifiable genetic etiologies. Spondylocostal dysostosis patients have a loss of normal vertebral morphology throughout the entire spine and have been shown to have mutations in delta-like 1 (DLLS), mesoderm posterior 2 (MESP2), and LFNG. Mutations in these affect the Notch signaling pathway and highlight the importance of Notch in vertebral column formation in humans (3, 41, 42). Spondylothoracic dysostosis (commonly referred to as Jarcho-Levin syndrome) is a confusing array of CVMs, all of which have rib anomalies with fusion of all ribs. A homozygous recessive nonsense mutation in MESP2 has been identified. Alagille syndrome is an autosomal dominant condition with bile duct paucity, cardiac, eye, kidney, pancreas, and vertebral anomalies in 22% to 87% of affected individuals. Mutations in JAG1 and NOTCH2 have been found in these patients (2, 3). Klippel-Feil syndrome was first reported in 1894 (43) and described in 1912 (44). Most cases have been found to be sporadic in families, but autosomal dominant, recessive, and X-linked forms have been reported. There has been recent evidence that PAX1 mutation and notch pathway mutations can be found in patients with Klippel-Feil syndrome (8, 45, 46 and 47). As isolated CVM are often sporadic occurrences with a given family, candidate gene analysis has been used. Using mouse-human synteny analysis, 27 eligible loci have been identified, of which 21 cause vertebral malformations in the mouse. Six of these—PAX1, WNT3A, DLL3, SLC35A3, T(Brachyury), and TBX6—have been studied in some detail and show promise as potential loci for the formation of CVMs in humans (3, 13, 14, 46, 47 and 48).
ASSOCIATED CONDITIONS
CVMs do not occur in isolation. As they develop during a critical stage of organogenesis, as many as 61% have other associated malformations (49), and the development of progressive deformity may lead to secondary organ involvement. The malformations seen are in general neurologic and visceral, with most tied to the level of the CVM.
Neurologic abnormalities have been described in 18% to 38% of patients with CVM (50, 51, 52, 53 and 54). A prospective evaluation of consecutive patients with CVM found that 38% had an intraspinal anomaly, with higher concentrations among patients with cervical and thoracic CVM, mixed patterns of segmentation and formation, and congenital kyphosis (50). Half of these patients had physical findings to suggest that they had an intraspinal lesion. Diastematomyelia was the most common finding followed by intraspinal lipoma, tethered cord, Chiari malformation with or without associated syringomyelia, dermoid cyst, and epidermoid cyst (Figs. 18-2, 18-3 and 18-4). For patients with an isolated hemivertebrae, there may also be a high incidence of intraspinal anomalies. Belmont found that of their 76 patients with C3VM, 29 had an isolated hemivertebrae, and of these, 8 (28%) had an MRI abnormality (55). Thirteen had abnormal clinical findings. A review of these patients and published series of similar patients suggests that an abnormal finding on the history of physical examination had an accuracy of 65%, sensitivity of 59%, specificity of 87%, a negative predictive value of 72%, and a positive predictive value of 74% (45, 50, 51, 56, 57).
For patients with known congenital cardiac abnormalities, the incidence of scoliosis has been found to be as high as 10% (58), but most of these are normally segmented (26, 58). Farley found that only 11/48 children (23%) with congenital heart disease (CHD) who had scoliosis had congenital scoliosis. The converse relationship of a higher incidence of CHD (baseline incidence 0.5% in the general population) in patients primarily found to have a congenital scoliosis has not been demonstrated, but it is believed that there is a higher incidence. Basu et al. (50) found cardiac abnormalities in 26% of patients with CVM. Two-thirds of these had knowledge of the cardiac abnormality before they presented for evaluation of their spine. Of those who did not have prior knowledge, of the cardiac defect, 2/10 (20%) did proceed to active management.
The incidence of renal anomalies in CVM has been reported as 26% to 37% (49, 50, 54, 59, 60 and 61). The most common anomalies have been unilateral renal agenesis, urinary duplication, ectopic kidney, reflux, and a horseshoe kidney. The incidence of renal anomalies may be higher if there is an associated rib anomaly in addition to the CVM (52). In most of the series, the renal anomalies were unsuspected, leading to general recommendations to screen children for these problems.
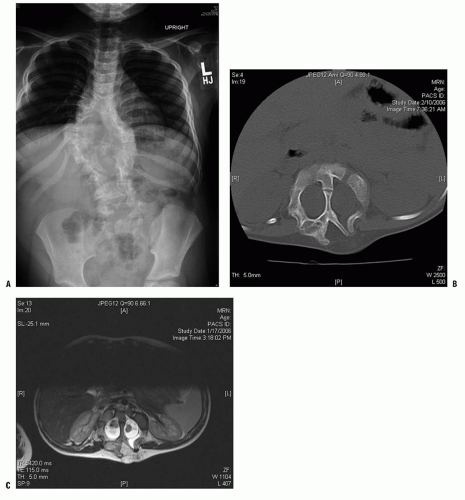 FIGURE 18-2. A: Example of diastematomyelia—plain radiographs may reveal a central canal bony process as the child matures. B: Plain radiographs. C: MRI findings. |
Morbidity associated with large progressive increases in deformity has largely been related to the development of restrictive lung disease and is believed to be a function of both altered alveolar multiplication and altered dynamics of breathing due to constriction of the thorax (62, 63, 64, 65, 66, 67, 68, 69, 70, 71 and 72). As compared to idiopathic scoliosis, the loss in vital capacity for a given Cobb angle is believed to be 15% greater for individuals with CVM (73). Several series have shown a progressive loss in vital capacity for progressive curves but have shown that if there is no progression and no associated severe rib anomalies, the vital capacity average for individuals with CVM is 87% of predicted (67, 69, 73, 74 and 75). Similarly, diffusion capacity for nonprogressive CVM has been found to be normal, suggesting that congenital scoliosis without fused ribs may not be associated with pulmonary hypoplasia or thoracic insufficiency syndrome (TIS) (72). Overall, however, the greater the deformity and the more proximal the deformity, the greater the apparent long-term impact upon spirometry (62, 63, 66, 69). For severe progressive curves, body mass index (BMI) less than the 5th percentile can be common, and this may create a higher risk situation for the operative management of these children. One of the primary clinical correlates to measurable declines in functional vital capacity
(FVC) in CVM is an altered BMI (74). Another is increased asymmetry of ventilation/perfusion (V/Q), which occurs in about 50% of children with TIS and congenital scoliosis (75). Coupled with this is the observation that diffusion capacity does decline with worsening deformity presumably due to a reduction in the total alveolar surface area (72). Cobb angle has not been correlated to either FVC changes or to altered function as measured by V/Q scans (68, 71). Surgical interventions aimed at stabilizing the chest/spine in patients with TIS have been shown to preserve growth-related changes in vital capacity over time but do not appear to be able to catch up for lost function (68, 70).
(FVC) in CVM is an altered BMI (74). Another is increased asymmetry of ventilation/perfusion (V/Q), which occurs in about 50% of children with TIS and congenital scoliosis (75). Coupled with this is the observation that diffusion capacity does decline with worsening deformity presumably due to a reduction in the total alveolar surface area (72). Cobb angle has not been correlated to either FVC changes or to altered function as measured by V/Q scans (68, 71). Surgical interventions aimed at stabilizing the chest/spine in patients with TIS have been shown to preserve growth-related changes in vital capacity over time but do not appear to be able to catch up for lost function (68, 70).
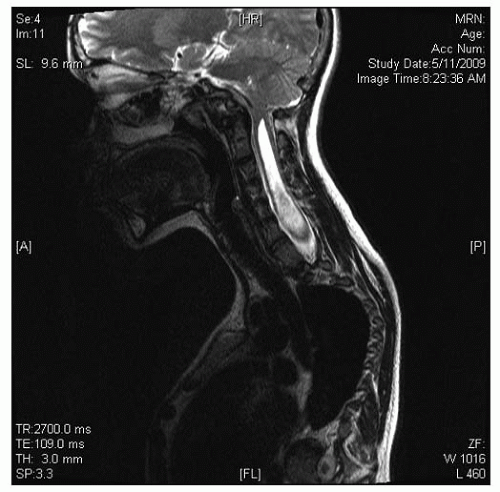 FIGURE 18-3. Example of syrinx with Chiari I malformation. |
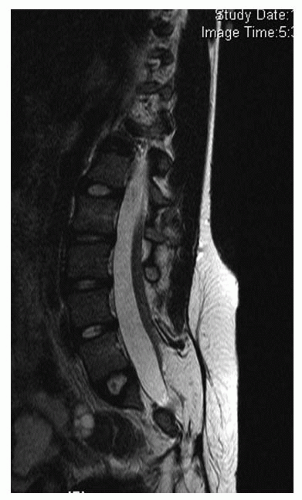 FIGURE 18-4. Example of a tethered spinal cord with an intraspinal lipoma. |
CLASSIFICATION/NATURAL HISTORY
Early reports of CVM suggested a benign natural history (76), but subsequent reports by Winter and Moe (77) and MacEwen et al. (78) identified severe deformities from high-risk patterns of deformity. Subsequent classifications for CVM have been several (77, 79, 80, 81, 82 and 83). Most have focused upon trying to assign a risk of progression by study of the natural history of deformities presenting to surgeons. Despite new knowledge gained by three-dimensional (3D) imaging using computed tomographic (CT) scans (79) and information about genetic mechanisms by which CVMs may form, the classification schemes proposed by Winter and Moe (77) and Nasca et al. (82) and modified by McMaster and Ohtsuka (80) remains the most useful and predictive (Fig. 18-5). The system defines malformations as either failures of formation, failures of segmentation, or combinations of these. The malformations can occur in frontal, sagittal, or both planes. The likelihood of progression is predicted by the anticipated potential for either unbalanced growth created by uneven growth potential due to failures of formation (hemivertebrae, wedge vertebrae) or growth retardation due to tethering created by failures of segmentation (bar). Approximately 10% of patients have patterns of CVM that are not definable by this classification scheme. The highest risk of progression was shown by McMaster to be in situations where there is unbalanced growth opposite an asymmetric tether as in a unilateral unsegmented bar with a contralateral hemivertebrae. The average annual rate of progression was found to be from 5 degrees to >10 degrees per year. The next highest risk of progression was in those with a unilateral unsegmented bar followed by double hemivertebrae (80) (Fig. 18-6). Reports of untreated patients followed in surgical clinics document a risk of progression to curves >40 degrees at maturity from 37% to 84% (76, 77, 80). For any given deformity type, there is variability in progression that may be due to the fact that it is difficult to account for variation in anterior and posterior CVMs at any given level. The most rapid times of progression were in the first 5 years of life and from ages 10 to 14 years. If there were clinically evident deformities in the first year of life, a worse overall prognosis was found (84, 85). The risk of progression has been predicated upon the belief that there is no growth potential of involved segments with CVM, leading to deformity and a shortened trunk (85). Campbell and Hell-Vocke (86) have questioned this with evidence of growth in CVM segments used forced growth techniques.
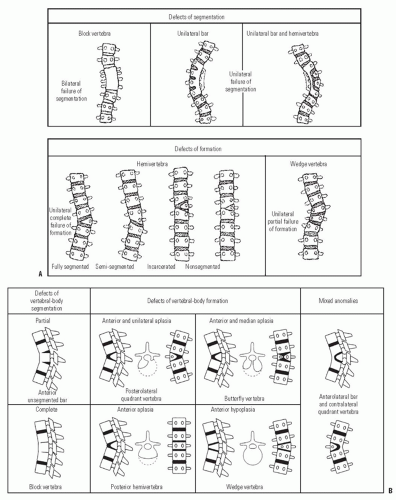 FIGURE 18-5. A: Classification of congenital scoliosis showing failures of formation and segmentation. Incarcerated hemivertebrae have compensatory deformities proximal and distal to it. The highest risk for progression lies with a hemivertebrae opposite a unilateral bar. B: Classification of congenital kyphosis. A high percentage of true congenital kyphosis deformities will progress and can lead to paraplegia especially if in the thoracic spine. |
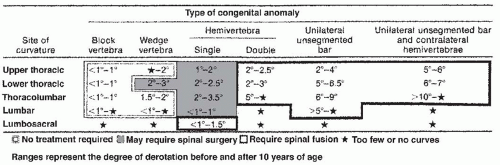 FIGURE 18-6. Risk of progression of types of CVM as compiled by McMaster. Highest rates of progression were in unilateral unsegmented bar and contralateral hemivertebrae. |
The clinical impact of progressive deformities associated with CVM has focused historically on respiratory consequences and more recently upon quality-of-life measures. There is no clear association that has been established between classifications of CVM and mortality or morbidity. Patients with severe progressive deformities have been demonstrated to have severe restrictive lung disease and significant morbidity with early death (62, 63 and 64, 85, 86). They have also been found to have a significantly diminished quality of life as measured by the Child Health Questionnaire in the domains of physical limitations and caregiver burdens, but not in psychosocial domains (87). Surgical intervention for these deformities has not yet been shown to alter these natural histories. It appears that pulmonary function decline can be stabilized with surgical treatment in some cases, but improvement in depressed vital capacity has not been observed (68, 70). Similarly, diminished quality of life does not appear to be improved after intervention (88).
EVALUATION OF PATIENTS WITH CVM
The wide range of presentations and the high incidence of visceral anomalies that can be present in patients with CVMs mandate that all evaluations include a careful history and physical examination. Presentations will range from severely involved children with obvious visceral and structural anomalies to those with nonprogressive and asymptomatic malformations.
Neurologic.
Many relatively uninvolved children will be referred due to incidental findings on imaging studies obtained for other reasons. Growth charts should be reviewed as children with CVMs may have altered growth velocities and can present with disproportionate growth retardation. Delays in developmental milestones such as walking and running and potty training can be important signs of an underlying spinal dysraphism as can symptoms of back pain in very young children. Physical findings of posterior midline trunk hairy patches, large nevi, or hemangiomas, atrophy of extremities, a cavus foot (Figs. 18-7 and 18-8), or any neurologic abnormalities (weakness, balance difficulties, sensory, hyperreflexia, asymmetric abdominal reflexes) can indicate an underlying spinal dysraphism in 50% to 80% of cases (19, 50, 51). It is widely recommended that an MRI of the neural axis be obtained prior to surgical intervention for CVM (45, 50, 51, 52 and 53, 56, 78, 89, 90 and 91). Less clear is whether there is benefit to the routine ordering of spine MRI scans for asymptomatic children with CVM who are not to undergo surgery. The positive and negative predictive value of physical examination and clinical history is 74% and 72% respectively for determination of MRI abnormalities (45, 50). As many younger children will require an anesthetic for a spine MRI, in asymptomatic nonprogressive deformities in children, observation to an older age is recommended. Intrauterine MRI has been tested for defining neural axis lesions in CVM. It has been found to be better than intrauterine ultrasound, but its overall accuracy and role in managing CVM are as yet unknown (92, 93, 94 and 95). For neonates with a suspected abnormality of the neural axis, ultrasound has been shown to be an effective screening tool and can be used
in a cost-effective manner depending upon the expertise of the individual performing it (8, 47, 54).
in a cost-effective manner depending upon the expertise of the individual performing it (8, 47, 54).
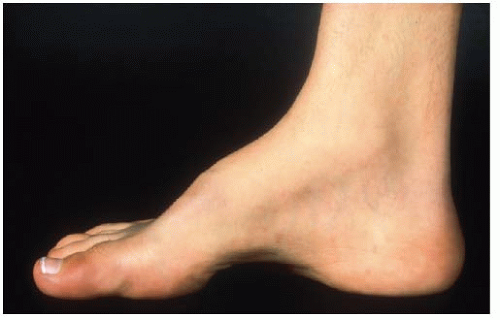 FIGURE 18-7. Cavus foot. High incidence of neurologic abnormalities associated with this finding. |
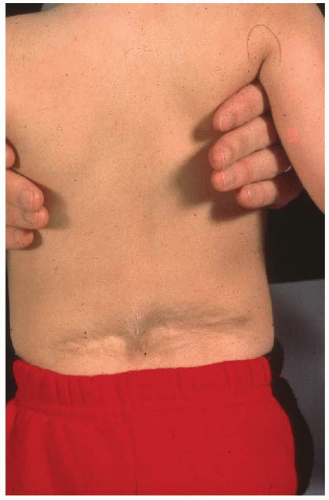 FIGURE 18-8. Midline hairy patch consistent with an underlying spinal dysraphism. |
Renal.
As up to one-third of patients may have an underlying renal anomaly and many will be asymptomatic, evaluation of the urinary system is generally advised (29, 49, 50, 52, 59, 69). Renal ultrasound has replaced contrast intravenous pyelogram as the imaging study of choice (60, 89). For patients who are undergoing MRI for evaluation of the neural axis, renal anomalies can be well-visualized when a modified spine MRI is ordered (96).
Cardiac.
As many as two-thirds of patients with cardiac abnormalities and CVM will have had diagnosis made by their primary care provider or a cardiologist prior to presentation for evaluation of their CVM (50, 58). For those who have not had prior evaluation and who are asymptomatic, the majority of the cardiac anomalies that may be present will not require active treatment. Cardiac screening is recommended for patients who are to undergo surgical treatment (89).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


