Congenital Deformities
Carl R. Wagreich
The purpose of this chapter is to present certain congenital deformities of the foot. Areas of discussion include epidemiology, genetic factors, pertinent classification systems, and various modes of conservative and surgical treatment. The conditions addressed include polydactyly, syndactyly, macrodactyly, cleft foot deformity, congenital overlapping toes exemplified by digiti quinti varus, underlapping or varus rotated lesser toes, hallux abductus interphalangeus, and congenital hemihypertrophy.
POLYDACTYLY
Polydactyly, defined as the presence of one or more extra or supernumerary digits or metatarsals of the feet or metacarpals of the or hands, is often described as one of the most common congenital deformities in these anatomic areas (1, 2, 3). Polydactyly has been reported throughout history. Within the Bible, in the Book of Samuel, 21:22, a giant with polydactyly is mentioned: “There was a man of great stature, that had on every hand six fingers, on every foot six toes.” In an ancient Arabian tribe, the Hyabites, polydactyly was so common that a child born with a normal compliment of digits was thought to be a product of adultery and was put to death (4).
Etiology
The origin of polydactyly appears to be genetic. Thirty-nine percent of patients’ charts that Venn-Watson reviewed noted a positive family history of polydactyly (3). However, he believed that the actual incidence of genetic association would have been higher had the history not been omitted in many of the charts. Several patients described distant relatives with similar conditions, and this finding led Venn-Watson to conclude that there was an incomplete penetrance. In other patients, he believed that polydactyly was possibly the result of a mutant gene. Phelps and Grogan found a positive family history of polydactyly in 30% of their patients (4). Watanabe et al. noted a familial incidence in only 8% of their patients (5). Their explanation of the disparate pattern when compared with studies in the United States was that polydactyly in the Asian population was possibly derived from other sources.
Temtamy and McKusick proposed that one form of polydactyly (postaxial type A) was inherited as a dominant trait with marked penetrance, whereas other forms were noted to be of more complex genetic types. Various other hand deformities were also discussed, most transmitted through autosomal dominant inheritance (6). When found in conjunction with other malformation syndromes, these conditions are thought to be the result of autosomal recessive inheritance (7,8).
No specific sex predilection has been reported (3, 4, 5,9,10). Bilateral involvement is seen in 25% to 50% of patients (3, 4, 5,10), and the incidence of polydactyly appears to be higher in black and Asian populations (9,11). In a large study involving 120,127 live births, Frazier listed an incidence of 3.6 in 1,000 live births in the black population and 0.3 in 1,000 live births in the white population (11). Woolf and Myrianthopolous reported the incidence of polydactyly to be 1.3 in 1,000 live births in the white population and 13.9 in 1,000 live births in the black population (12).
Classification
Various systems for classification of polydactyly have emerged over the years (3, 4, 5,13,14). Temtamy and McKusick differentiated medial ray (preaxial) polydactyly from lateral ray (postaxial) polydactyly and mixed forms (13). This basic nomenclature has been accepted, although several modifications and expansions to their classification system have been offered.
Postaxial Polydactyly
Postaxial polydactyly, the most common presentation of this condition, accounts for 79% to 86% of reported cases
in larger studies (4,5). Postaxial polydactyly was further differentiated by Tetamy and McKusick into types A and B. Type A represents a fully developed accessory digit that articulates with either the fifth metatarsal or metacarpal or with a duplicated fifth metatarsal or metacarpal. In type B, the accessory digit is devoid of osseous components and frequently manifests as only a skin tag (13). However, this basic classification system failed to distinguish the wide range of intermediate forms of postaxial polydactyly. Venn-Watson further divided postaxial polydactyly into five specific morphologic patterns, based on the degree of metatarsal duplication (3). From the least differentiated to the most differentiated, they are as follows: soft tissue duplication, wide metatarsal head, T-metatarsal, Y-metatarsal, and complete duplication (Fig. 1). Phelps and Grogan noted that the most common form of postaxial polydactyly found in their patients was duplication of the proximal phalanx with either a block or a wide metatarsal head (4).
in larger studies (4,5). Postaxial polydactyly was further differentiated by Tetamy and McKusick into types A and B. Type A represents a fully developed accessory digit that articulates with either the fifth metatarsal or metacarpal or with a duplicated fifth metatarsal or metacarpal. In type B, the accessory digit is devoid of osseous components and frequently manifests as only a skin tag (13). However, this basic classification system failed to distinguish the wide range of intermediate forms of postaxial polydactyly. Venn-Watson further divided postaxial polydactyly into five specific morphologic patterns, based on the degree of metatarsal duplication (3). From the least differentiated to the most differentiated, they are as follows: soft tissue duplication, wide metatarsal head, T-metatarsal, Y-metatarsal, and complete duplication (Fig. 1). Phelps and Grogan noted that the most common form of postaxial polydactyly found in their patients was duplication of the proximal phalanx with either a block or a wide metatarsal head (4).
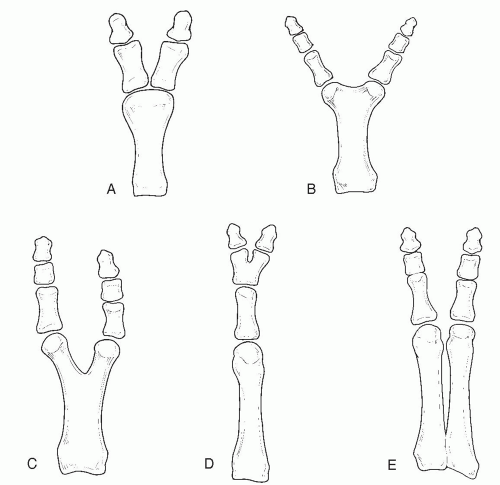 FIG. 1. Postaxial polydactyly, modified from the Venn-Watson classification. A: Wide metatarsal head. B: T-shaped metatarsal. C: Y-shaped metatarsal. D: Partial polydactyly. E: Complete duplication. |
Watanabe et al. compiled the most extensive study of polydactyly of the foot that included 265 individual cases (5). These investigators provided an even more elaborate classification system, with postaxial polydactyly divided into fifth and sixth ray duplication, each with subtypes. Fifth ray duplication accounted for 76% of all postaxial polydactyly. Fifth ray polydactyly was accompanied by a 3% incidence of associated anomalies, and a 35% incidence of associated anomalies was seen in patients with sixth ray duplication.
Preaxial Polydactyly
Preaxial polydactyly of the foot is seen in 8% to 15% of patients (4,5). Temtamy and McKusick described four subtypes: type 1, duplication of the first digit; type 2, polydactyly of a triphalangeal first digit; type 3, polydactyly of the second digit; and type 4, polysyndactyly (13). Venn-Watson
described variants consisting of a short block metatarsal or a wide metatarsal head (3). Phelps and Grogan noted that the most common configuration was duplication at the proximal phalangeal level with a block first metatarsal (4) (Fig. 2).
described variants consisting of a short block metatarsal or a wide metatarsal head (3). Phelps and Grogan noted that the most common configuration was duplication at the proximal phalangeal level with a block first metatarsal (4) (Fig. 2).
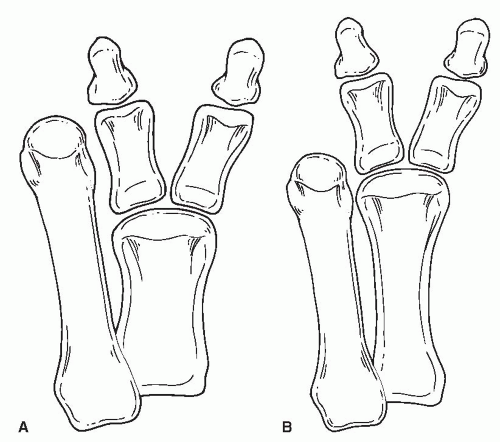 FIG. 2. Preaxial polydactyly, modified from the Venn-Watson classification. A: Short block first metatarsal. B: Wide metatarsal head. |
Watanabe et al. proposed four groups: tarsal, metatarsal (with three subtypes), proximal phalangeal (with five subtypes), and distal phalangeal (with six subtypes) (5). Metatarsal and proximal phalangeal types were most commonly noted, each accounting for 36% of preaxial polydactyly. These authors postulated that the tarsal type of medial ray duplication represented the true prehallux. It was seen in only one patient, with the duplicated digit articulating with the navicular. Associated anomalies including deformity syndromes were found in 59% of the patients with preaxial polydactyly, frequently involving the hand.
Central Ray Polydactyly
Duplication involving the central rays has been reported in approximately 6% of patients presenting with polydactyly (4,5). Phelps and Grogan noted that this most often consisted of a hypoplastic metatarsal (4). Watanabe et al. divided this condition into four types: metatarsal, proximal phalangeal, middle phalangeal (divided into two subtypes), and distal phalangeal (5). All patients except one had duplication of the second toe. The most common level of duplication was the distal phalangeal type. Associated anomalies were found in 20% of patients with central ray polydactyly.
Associated Syndromes
Polydactyly may be associated with other syndromes. Therefore, when a child is born with polydactyly, it may not be an isolated deformity. Opinions vary regarding whether preaxial polydactyly or postaxial polydactyly is accompanied by a higher incidence of associated congenital anomalies. According to Castle, preaxial polydactyly is usually an isolated deformity and is rarely associated with another syndrome. The most common associated syndrome seen with polydactyly of the foot is polydactyly of the hand (4,10). Syndactylism of the toes was the second most common associated syndrome, found in 22% of patients with polydactyly of the foot (4). Postaxial polydactyly has been more commonly associated with genetic syndromes (7). Conversely, Watanabe et al., in their study, found a 59% incidence of associated congenital anomalies in preaxial polydactyly as opposed to a 38% incidence in postaxial polydactyly (5).
One set of genetic syndromes that include polydactyly comprises the short rib polydactyly syndromes. These disorders are a group of lethal syndromes in which the child usually does not survive through infancy (15). In 1992, the International Working Group on Constitutional Diseases of Bone recognized six types of short rib polydactyly syndromes. They include type I (Saldino-Noonan), type II (Majewski), type III (Verma-Naumoff), type IV (Breemer-Langer), plus asphyxiating thoracic dysplasia, and Ellis-van Creveld dysplasia (16). Skeletal dysplasias include extremely short ribs, ovoid vertebral bodies, an irregularly shaped pelvis, horizontal acetabula, short long bones, and premature ossification of distal femoral epiphyses. The skeletal abnormalities accompany many visceral anomalies including cystic kidneys, liver, and pancreas, cleft lip and palate, esophageal and anal atresia, and congenital heart defects (7,15, 16, 17, 18, 19). Apert syndrome is similar to the short rib polydactyly syndromes in skeletal and visceral abnormalities. However, polydactyly
is not as commonly seen in Apert syndrome. In addition, craniofacial abnormalities with varying degrees of mental retardation have been noted (20,21).
is not as commonly seen in Apert syndrome. In addition, craniofacial abnormalities with varying degrees of mental retardation have been noted (20,21).
Polydactyly has been associated with atrioventricular septal defect (22,23), fibular dimelia, absent tibiae (24, 25, 26, 27), syndactyly (28, 29, 30, 31), talipes equinovarus (28), hydrolethalus syndrome (32,33), hydrometrocolpos (34), drug-related fetal hydantoin syndrome (35), Proteus syndrome (36), and a host of newly reported syndromes. Because many deformity syndromes are poorly defined, any variability in the components that are reported may at times result in the description of a new syndrome (29,30,37,38).
Treatment
Conservative treatment is not routinely practiced in the symptomatic child with polydactyly. In many instances, duplication of the part makes shoe fitting and subsequent function difficult. Parents are often concerned about future potential functional impairment as well as cosmetic appearance (3). Because the deformity has both physical and emotional sequelae, treatment of polydactyly is usually surgical (1,39). Conservative measures that may be used include padding, strapping, orthodigital devices, wider shoes, extra-depth shoes, and orthotic devices (9,40). Generally speaking, these measures provide only temporary relief.
Treatment of postaxial polydactyly, type B (rudimentary digit only) may be initiated shortly after birth once the neonate’s condition is stable. The treatment includes tying off the rudimentary digit with a ligature suture in the nursery (41). No reports of exsanguination have been reported. However, this procedure is generally not performed if osseous components are present in the digit. If skeletal components are present, it is generally preferable to wait until the child is a few months old before a surgical procedure is performed, to ensure complete excision of all accessory osseous structures (42).
Opinions on the timing of surgical intervention vary. Chiang and Huang reported an age range at the time of surgery between 4 months and 13 years and 4 months, with a mean age of 3 years and 3 months (1). Phelps and Grogan reported an average age of 3.8 years but noted that an age of approximately 1 year was preferred (4). Watanabe et al. reported a range from 3 months to 5 years with an average of 22 months in preaxial polydactyly, 36 months in central ray polydactyly, and 26 months in postaxial polydactyly (5). According to Tachdjian, the optimum age for surgical treatment is between 9 months and 1 year (43). Other investigators have advocated surgical treatment before the child’s initiation of weight bearing, to allow better shoe fitting (3). DeValentine noted that surgery for polydactyly should generally be delayed until the child is at least 1 year of age. Anesthesia is better tolerated at that point, and osseous development is more advanced, a feature that, in turn, allows for better surgical planning and facilitates the operation because of the larger size of the foot. In some cases, phalangeal and metatarsal ossification may not be adequate to determine which toe should be removed until the patient is 2 to 3 years of age. Surgical treatment is generally preferred before the child starts school, to avoid problems with shoe wear (42).
Surgical Considerations
The preoperative evaluation of the patient with polydactyly should, most importantly, take into account the central metabolic and genetic status. As previously discussed, the factor of other disease states may need to be investigated. Standard radiographs are often useful in assessing the level and severity of the pathologic anatomy. However, they may not be sufficient to render a complete picture of soft tissue anomalies that may be encountered at the time of surgery. Investigators have suggested that computed tomography scans and magnetic resonance imaging studies may be helpful in preoperative planning (2,40). However, besides being costly, these tests may require significant levels of patient sedation to obtain a valid study.
Each case of polydactyly should be considered individually. The goals of surgery in polydactyly are to remove anomalous tissue and to restore function (39,44, 45, 46). Skin incisions over bony prominences are preferably avoided to prevent the formation of scar tissue over pressure areas. Furthermore, the incision should be planned to create equal length and shape to the wound, to promote good plastic skin closure. Resecting bone at the junction of the anomaly reduces the risk of cortical splitting.
In most patients with preaxial polydactyly, the most medial duplicated metatarsal and digit are excised, whereas in most cases of postaxial polydactyly, the most lateral duplicated metatarsal and digit are excised (4,9,47). The exception to this rule is when the most lateral or medial duplicated digit is preferred cosmetically or would provide better function. Then, alternatively, the innermost duplicated digit is excised (3,4,42). This excision, unfortunately, tends to leave a space between the remaining digits and a wider foot. Under
these circumstances, after removal of the inner metatarsal and digit, osteotomy of the most medial metatarsal in a preaxial deformity and of the most lateral metatarsal in a postaxial deformity may be undertaken to narrow the foot (39,45) (Figs. 3, 4, 5 and 6).
these circumstances, after removal of the inner metatarsal and digit, osteotomy of the most medial metatarsal in a preaxial deformity and of the most lateral metatarsal in a postaxial deformity may be undertaken to narrow the foot (39,45) (Figs. 3, 4, 5 and 6).
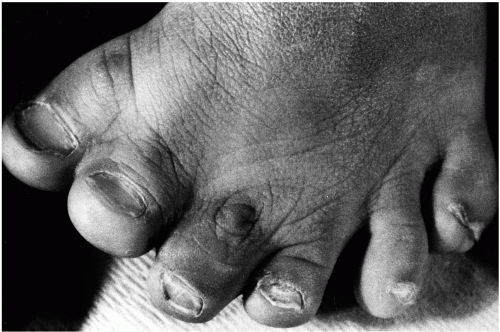 FIG. 3. Clinical appearance of a patient with preaxial polydactyly. |
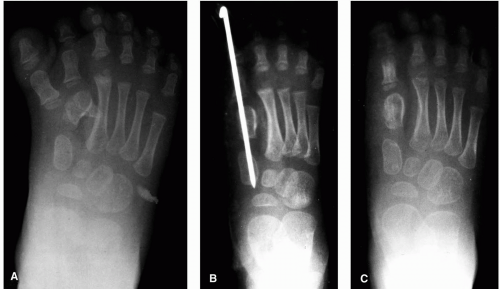 FIG. 4. A: Radiograph of a patient with preaxial polydactyly with complete duplication of the hallux and a block metatarsal. B: Postoperative radiograph after removal of the medial duplicated digit with Kirschner wire stabilization. C: Radiograph 6 weeks after surgery. |
Excision of a digit is often performed through a rackettype incision. If the metatarsal head is widened, then it may be narrowed to a more normal width. Resection through the physis has not been shown to result in premature closure of the growth plate (3,4). Any duplication of the metatarsal is removed flush with the remaining bone. If complete duplication of a metatarsal is present, then a ray resection may be preferred. With excision of a central ray duplication, the intermetatarsal ligaments may be reconstructed to counteract splaying of the forefoot. However, residual forefoot splaying has been noted to persist in most patients despite this technique (4). Fixation devices such as pins, staples, and casts may provide added stability to the remaining segments. Joint
incongruity is frequently noted to persist after resection. If they are properly stabilized postoperatively, these articular surfaces remodel, especially in the young child. In addition, any residual bowing of the metatarsal after resection of the lateral portion of the duplicated Y-shaped metatarsal remodels (3).
incongruity is frequently noted to persist after resection. If they are properly stabilized postoperatively, these articular surfaces remodel, especially in the young child. In addition, any residual bowing of the metatarsal after resection of the lateral portion of the duplicated Y-shaped metatarsal remodels (3).
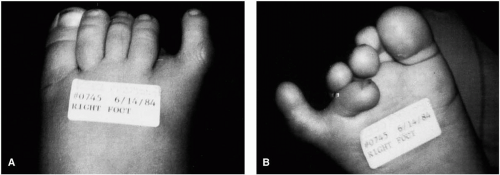 FIG. 5. Repair of postaxial polydactyly with duplication of the fifth digit and a wide metatarsal head. Preoperative appearance of the foot from the dorsal view (A) and the plantar view (B). On the dorsal view, the fifth toe is not visible because of the extreme plantar adducto varus rotation. C: Preoperative radiograph. D: Two converging semielliptic incisions are used around the most lateral digit. The extensor and flexor tendons are severed proximally. The wide metatarsal head before (E) and after (F) remodeling. G: The deformity at the proximal interphalangeal joint is reduced and is maintained in position with a Kirschner wire. H: Appearance of the foot 7 weeks postoperatively. |
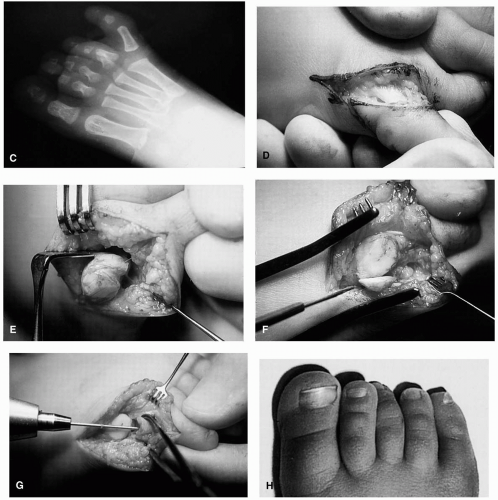 FIG. 5. Continued. |
The main complication associated with the repair of postaxial polydactyly is residual soft tissue or osseous bulk that impairs shoe fitting. Venn-Watson attributed this problem to an inadvertent shaving of a Y-shaped metatarsal head or when a duplicated metatarsal was left once the digit itself was removed while the child was in the nursery (3).
The most common complication associated with the repair of preaxial polydactyly in the report by Venn-Watson was
a residual hallux varus. Because of this complication, Venn-Watson recommended that the surgeon lengthen the abductor hallucis tendon and plicate the adductor tendon into the joint capsule. Casting was advocated to maintain position (3). Phelps and Grogan noted similar problems with hallux varus, which developed in 14 of 16 patients. The duplicated digit was removed and capsular tissues were reapproximated without addressing tendon structures (4). Pinning the joint for several weeks would probably be a more suitable method of splinting than casting while the soft tissues are healing (42). Other problems associated with this type of surgery have been increased tissue bulk and a short first metatarsal, the latter of which typically creates later problems with foot function (3,4,42).
a residual hallux varus. Because of this complication, Venn-Watson recommended that the surgeon lengthen the abductor hallucis tendon and plicate the adductor tendon into the joint capsule. Casting was advocated to maintain position (3). Phelps and Grogan noted similar problems with hallux varus, which developed in 14 of 16 patients. The duplicated digit was removed and capsular tissues were reapproximated without addressing tendon structures (4). Pinning the joint for several weeks would probably be a more suitable method of splinting than casting while the soft tissues are healing (42). Other problems associated with this type of surgery have been increased tissue bulk and a short first metatarsal, the latter of which typically creates later problems with foot function (3,4,42).
 FIG. 6. A: Central ray polydactyly with duplication of the second metatarsal and digit. B: Postoperative radiographs after removal of the duplicated medial second metatarsal and digit. |
SYNDACTYLY
Syndactyly is defined as a congenital or acquired deformity in which webbing persists between adjacent digits from birth or secondary to injury (48,49). Drinkwater provided one of the earliest reports of syndactyly with synphalangism in 1917 and traced the deformity to the time of King Henry VI of England. The first member of the family who was known to have the condition was killed in 1453. His tomb was opened in 1874, and his finger bones were shown to have the same bony ankylosis as those of his descendants (50).
Etiology
Syndactyly of the toes is one of the most frequently encountered congenital anomalies (51). It often involves the second and third toes in the foot (8,52). Investigators generally agree that syndactyly is caused by a rapid arrest of embryologic development from the sixth to eighth week of intrauterine life. Because the webbing between the second and third toes is the last to disappear, this area is the most sensitive to intrauterine insult (42,44,48,53). Genetic factors have most often been implicated as the source for this condition (13). Traumatic causes are usually secondary to burns. Syndactyly is ten times more common in whites than in blacks and presents in roughly equal numbers in bilateral and unilateral forms. Males and females are similarly affected, although Davis and German noted a greater male predominance of 68% (52). The incidence is about 1 in 2,500 to 3,000 live births (8,52).
Classification
Two classification systems for syndactyly are recognized. Davis and German divided the condition into four classes (52), as follows:
Incomplete: Webbing does not extend to the most distal aspect of the involved digits.
Complete: Webbing extends to the ends of the involved digits.
Simple: A soft tissue connection alone exists.
Complicated: The phalanges are abnormal in size, shape, number, or arrangement.
DeValentine noted that the complicated forms may share neurovascular, tendinous, or osseous structures. Furthermore, he noted that a fifth class, complicated-complex, was used by some investigators. This class consists of three or more digits with interposed incomplete structures (42).
Temtamy and McKusick described a classification based on associated syndromes and deformities (13). Two classes were described: syndactyly and syndromatic syndactyly. Isolated syndactyly is divided into five phenotypic types that are all inherited as autosomal dominant disorders. They are as follows:
Type 1. Zygodactyly Webbing is usually present between the third and fourth fingers. In the foot, it usually involves the second and third toes. Syndactyly may be partial or complete. These investigators also noted that frequently one could see some degree of webbing between the second and third toes, but this was not necessarily related to type 1.
Type 2. Synpolydactyly Syndactyly of the third and fourth fingers is present, with polydactyly of components of the fourth finger in the web. In the foot, one would see polydactyly of the fifth toe included in the web between the fourth and fifth digits.
Type 3. Ring finger-small finger syndactyly This type manifests with complete syndactyly between the fourth and fifth fingers, typically bilaterally. The feet are not affected.
Type 4. Hass type This type involves complete syndactyly of all fingers. Occasionally, patients have a sixth metatarsal and phalanx. No foot involvement occurs.
Type 5. Syndactyly with metacarpal and metatarsal fusion There is syndactyly of the third and fourth fingers and the second and third toes. Associated findings are fusion of the fourth and fifth metacarpals and metatarsals and occasionally the third and fourth metatarsals.
Syndromatic syndactyly is divided into two groups: the first comprises syndromes in which syndactyly is predominant, and in the second group, syndactyly is secondary to other medical problems. The list of associated syndromes and deformities is voluminous. A partial list includes split hand-split foot (Czeizel-Losonci syndrome) (54), talipes equinovarus (28), tibial hypoplasia or aplasia, polydactyly (30), occipitocervical encephalocele, vertebral fusion (29), cleft palate, Apert syndrome (55), renal and anogenital malformations (56), mental retardation (57), dysplasia epiphysealis hemimelica (58), cardiac conduction deficits (59), amniotic band syndrome (60), and Fraser syndrome (crypto-phthalmos, hidden eye syndrome) (61).
Treatment
The definitive treatment of syndactyly is surgery. Virtually all reports in the literature represent syndactyly as a purely cosmetic problem, yet the emotional sequelae of the deformity are recognized (31,44). Some authors believe that because the problem is purely cosmetic, it should not be repaired surgically (43,62). However, some reports have noted syndactyly involving the hallux and second digit that resulted in the use of altered gait patterns to avoid pain (49).
Surgery is generally performed in the pediatric patient after 1 year of age because the results in younger children are generally poor. At this earlier age, the commissures tend to close and advance. Children older than 5 years of age are subject to ridicule in school. Therefore, investigators have recommended that surgical correction be performed when patients are between the ages of 2 and 4 years (8). DeValentine recommended waiting until adolescence so the child could participate in the decision and better assist in the postoperative care. However, he also indicated that more complex cases could require earlier intervention, generally between the ages of 1 and 5 years. Preference was expressed for the upper limit of this range, so a more complete radiographic assessment and better intraoperative visualization of anatomic structures could be available (42).
As in the patient with polydactyly, one should consider the general metabolic and genetic status of the patient and the potential for other disease states. Standard radiographs are typically made preoperatively to evaluate for the presence of synphalangism.
Surgical Considerations
The goals of surgery for syndactyly are to provide adequate soft tissue coverage to adjacent toes, to prevent contractures secondary to the surgical procedure, and to create a commissure or space at the base of the digits (49). Various considerations are important in planning a procedure that will achieve these goals. Vascular compromise may occur, and to avoid excessive tension on the desyndactylized digits, skin grafting may be necessary. Curved or zig-zag incisions may result in less digital contracture. After suture removal, continuous-pressure taping may be applied to the web space for up to 2 months to reduce “web creep” (51).
Three types of surgical procedures are used in desyndactylization. They include flaps, grafts, and tissue expansion. Flaps may take on a number of configurations for desyndactylization. Itoh and Arai described proximal zig-zag lines on the dorsum that are made parallel to the metatarsophalangeal joint axis (63) (Fig. 7). Flaps C and D are made of dorsal skin because they will be under greater tension. A subcutaneous pentagonal pedicle flap of plantar skin is advanced to cover the base of the interdigital space. Coleman et al. discussed this technique in the foot (48). After the tracing of the dorsal flaps with a skin marker, 27-gauge needles were placed through the toes from dorsal to plantar to provide proper positioning for the apices of the plantar flaps. DeValentine similarly discussed this type of approach and noted that a
small area on each digit would require grafting in some instances (42).
small area on each digit would require grafting in some instances (42).
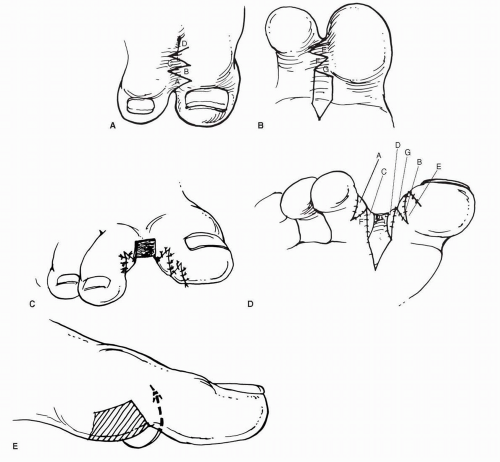 FIG. 7. Itoh method of desyndactylization. A,B: Proposed dorsal and plantar skin incisions with proximal advancement flap on the plantar side. C,D: After coverage of the interdigital spaces. E: Advancement of plantar flap to cover the central defect. |
Other authors have presented cases using the technique initially proposed by Didot in 1849 for desyndactyly of the fingers (49,64). Today, this procedure is rarely performed in the hand because of restrictive scar formation. However, the toes do not require the same degree of flexibility, and the procedure is much simpler than some of the alternative methods and provides excellent functional and cosmetic results in the foot (Fig. 8).
Grafting is also commonly practiced. Full-thickness grafts are recommended over split-thickness (defatted) grafts because the split-thickness grafts are more likely to contract and to deform the digits (8,65). Other investigators recommend full-thickness grafts in children, but they consider split-thickness grafts in adults and adolescents because full-thickness grafts require a greater blood supply for viability (42). Postoperative defects should be avoided because they also add to contracture formation (31). Donor sites may include the medial submalleolar region (65), the lateral submalleolar region (66), the dorsum of the foot (67), the groin (8), and the abdominal region (68). Tissue expanders have been used to reduce the need for skin grafts (68). This procedure allows local tissue to cover the soft tissue defects that are created. Furthermore, the tissue appears to be free from the pigmentation problems sometimes associated with skin grafts, and scarring and contracture are minimal. The disadvantages of this approach include the necessity for two operations, possible rupture or leakage of the expander, repeated injections, and a greater amount of time to complete the treatment.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


