Chapter 14 Complement and SLE
Historical Overview
The complement system is arguably linked more intimately to systemic lupus erythematosus (SLE) than to any other human disease. This association has been recognized for decades and, until recently, was viewed as inexplicably paradoxical. Two seemingly irreconcilable early observations formed the foundation for this conundrum. First, in 1951 Vaughan was the first to assay serum complement in cases of SLE.1 His team determined from four cases that a diminished total complement activity (CH50) value correlated with disease activity. Elliott confirmed this finding and noted complement depression to be particularly associated with “albuminuria.”2 Lange discovered that complement was diminished in virtually all cases of acute, but not in chronic, glomerulonephritis, and that low complement was characteristic of SLE whether or not there was renal involvement.3 Schur, in the largest study to that time, found CH50 to be below 50% of normal in 24% of patients with active SLE and in 46% of those with renal involvement, but in only 4% with inactive disease.4 Schur suggested that complement levels were of particular value in following and evaluating patients with SLE, especially those with nephritis. These seminal observations were followed by a large body of work from many laboratories demonstrating that complement activation, reflected by diminished serum levels of C3, C4, and diminished CH50, plays a major role in the tissue inflammation and organ damage that result from lupus pathogenesis.
Seemingly paradoxical to these findings was a second set of observations that demonstrated a strong association between hereditary homozygous deficiency of the classical pathway components and development of SLE.5 In fact, inherited complement deficiency is still recognized as conferring the greatest known risk for development of SLE. Thus, for decades this paradox has been pondered. How is it that complement deficiency may be causative in SLE yet activation of this same inflammatory cascade is detrimental in patients who already have the disease?
Biology of the Complement System
Investigation of complement originated in the late 19th century, when a heat-labile serum component with nonspecific activity (now known to be complement) was found capable of facilitating the killing of bacteria by a heat-stable serum component with antigen specificity (now known to be antibodies).6 Subsequently, similar heat-labile and heat-stable factors were demonstrated to mediate lysis of erythrocytes sensitized by immune sera. In 1899, the term complement was introduced by Paul Ehrlich to emphasize that the heat-labile factors present in fresh serum “complemented” the heat-stable specific factors mediating immune bacteriolysis and hemolysis.7 Although its name may imply an ancillary role in immunity, later studies have demonstrated that the complement system is not only a vital component of host defense, through participation in innate immune response and adaptive immunity, but also an “accidental” culprit in the pathogenesis of SLE and other immune-inflammatory diseases.
Protein biochemistry studies conducted in the late 1960s and throughout the 1970s and 1980s have greatly advanced our understanding of the biochemical nature of the complement system.8 The complement system comprises more than 30 plasma and membrane-bound proteins that form three distinct pathways (classical, alternative, and lectin) designed to protect against invading pathogens (Table 14-1 and Figure 14-1).9–11 Many of the complement proteins exist in plasma as functionally inactive zymogens until appropriate events trigger their activation. Once activated, the proteins within each pathway undergo a cascade of sequential serine protease–mediated cleavage events, release biologically active fragments, and self-assemble into multimolecular complexes. In general, activation of the complement system can be viewed as a two-stage process. The first stage, unique to each of the three activation pathways, involves the early complement components that lead to the formation of the C3 convertases. The second stage, common to all three pathways once they converge, results in the formation of activation products (such as C3a and C5a) and a lytic complex consisting of the terminal complement components (see Figure 14-1).
TABLE 14-1 Components of the Human Complement System
| Effector Protein | Function /Pathway Involved | Mr (kd) |
|---|---|---|
| C1q | Recognition, binding/classical | 450 (a six-subunit bundle) |
| C1r | Serine protease/classical | 85 |
| C1s | Serine protease/classical | 85 |
| C4* | Serine protease (C4b); anaphylatoxin (C4a)/classical | 205 (a 3-chain, αβγ, complex) |
| C2 | Serine protease (C2a); small fragment with kinin activity (C2b)/classical | 102 |
| C3† | Membrane binding, opsonization (C3b); anaphylatoxin (C3a)/terminal | 190 (a 2-chain, αβ, complex) |
| C5 | MAC component (C5b), anaphylatoxin (C5a)/terminal | 190 (a 2-chain, αβ, complex) |
| C6 | MAC component/terminal | 110 |
| C7 | MAC component/terminal | 100 |
| C8 | MAC component/terminal | 150 (a 3-chain, αβγ, complex) |
| C9 | MAC component/terminal | 70 |
| Factor B | Serine protease/alternative | 90 |
| Factor D | Serine protease/alternative | 24 |
| Properdin | Stabilizing of C3bBb complexes/alternative | 55 (monomers); 110, 165, 220, or higher (oligomers) |
| MBL | Recognition, binding/lectin | 200-400 (2-4 subunits with three 32 KD chains each) |
| MASP-1 | Serine protease/lectin | 100 |
| MASP-2 | Serine protease/lectin | 76 |
| Soluble Regulatory Protein | Function | Mr (kd) |
| C1 inhibitor (C1-INH) | Removal of activated C1r and C1s from the C1 complex | 105 |
| C4-binding protein (C4bp) | Binding to C4b and displacing C2a in the C4bC2a complex; accelerating decay of C3 convertase; cofactor for factor I | 570 (an 8-subunit complex) |
| Factor H | Displacing Bb in the C3bBb complex; cofactor for factor I | 160 |
| Factor I | Serine protease cleaving C3b and C4b | 88 |
| Clusterin | Preventing insertion of soluble C5b-9 complexes into cell membranes | 70 |
| S protein (vitronectin) | Preventing insertion of soluble C5b-9 complexes into cell membranes | 84 |
| Carboxypeptidase N | Inactivating anaphylatoxins | 280 (a multi-subunit complex) |
| Membrane-Bound Regulatory Protein | Function | Mr (kd) |
| CD35 (CR1) | Binding C3b and C4b; cofactor for factor I | 190-280 (four isoforms) |
| CD46 (MCP) | Promoting C3b and C4b inactivation by factor I | 45-70 (different glycosylation forms) |
| CD55 (DAF) | Accelerating decay of the C3bBb and C4b2a complexes | 70 |
| CD59 (protectin; H19) | Preventing C9 incorporation into the MAC in a homologous restriction manner | 18-20 |
| Complement Receptor | Structure/Mr (kd) | Complement Ligand(s)‡ |
| CR1 (CD35) | Single chain/190-280§ | C3b; C4b; iC3b; C1q |
| CR2 (CD21) | Single chain/140-145 | C3dg/C3d; iC3b; |
| CR3 (CD11b/CD18) | Two-chain, α/β/ 170/95 | iC3b |
| CR4 (CD11c/CD18) | Two-chain, α/β/150/95 | iC3b |
| cC1qR (calreticulin) | Single chain/60 | C1q (collagenous tail); MBL |
| gC1qR | Tetramer/33 per subunit | C1q (globular head) |
| C1qRP | Single chain/126 | C1q (collagenous tail) |
| C3a receptor | Single chain/50? | C3a |
| C5a receptor (CD88) | Single chain/50 | C5a |
DAF, decay-accelerating factor; kd, kilodalton; MAC, membrane attack complex; MASP, mannose-binding protein–associated serine protease; MBL, mannose-binding lectin; MCP, membrane cofactor protein; Mr, molecular mass (molecular weight); r, receptor.
* Serum concentration range considered normal: 20-50 mg/dL.
† Serum concentration range considered normal: 55-120 mg/dL.
‡ Noncomplement ligands (e.g., Epstein-Barr virus for CR2 and fibrinogen for CR3 and CR4) not listed.
§ Four allotypes with different numbers of SCR and displaying distinct Mr under reducing condition: CR1-A (220 kd), CR1-B (250 kd), CR1-C (190 d), and CR1-D (280 kd).
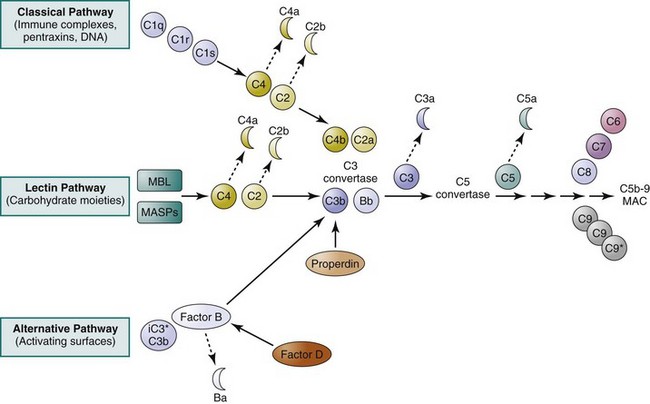
Complement Activation Pathways
The classical pathway of complement activation is thought to play an important role in SLE pathogenesis. There are five proteins specific to activation of the classical pathway: C1q, C1r, C1s, C4, and C2 (see Figure 14-1). Activation of this pathway begins when C1q binds to the Fc portion of immunoglobulin G (IgG) (particularly IgG1 and IgG3) or IgM molecules that are bound to an antigen. The binding of C1q to an antigen-antibody complex (immune complex) leads to activation of C1r (a serine protease), which, in turn, leads to activation of C1s (also a serine protease). C1s enzymatically cleaves the other two classical pathway proteins, C4 and C2, generating and releasing two small soluble polypeptides, C4a and C2b. At the same time, this proteolytic cleavage leads to the formation of a surface-bound bimolecular complex, C4b2a, which functions as an enzyme and is referred to as the classical pathway C3 convertase. Studies have now shown that in addition to immune complexes, molecules of the “pentraxin” family (such as C-reactive protein and serum amyloid P) and DNA have also been shown capable of directly interacting with C1q and thus initiating the classical pathway.11
Unlike activation of the classical pathway, activation of the alternative pathway does not depend on antibodies but can be triggered by carbohydrates, lipid, and proteins found on surfaces of microbial pathogens. Three plasma proteins are unique to the alternative pathway: factor B, factor D, and properdin (see Figure 14-1). Normally, native C3 molecules undergo a so-called C3 tickover process, a continuous, low-rate hydrolysis of the thioester group that generates iC3* (hydrolyzed C3) and, subsequently, C3b fragments.12 A fraction of these spontaneously generated C3b fragments may covalently attach to the surface of microbial pathogens and host cells via thioester bonds. The bound C3b molecules are capable of binding factor B. Once bound, factor B is cleaved into Ba and Bb fragments by factor D (a serine protease). While the small, soluble Ba fragment diffuses away from the activation site, the Bb fragment remains associated with C3b. Like the C4b2a complex in the classical pathway, the surface-bound C3bBb complex serves as the alternative pathway C3 convertase. The C3bBb complexes, if bound to host cells, are rapidly degraded by several regulatory proteins, thereby preventing self-damage. However, the C3bBb complexes associated with microbial pathogens are stabilized by the binding of properdin and prevented from being degraded by factor H and factor I. It has been suggested that properdin can also function as pattern-recognition molecules and initiate complement activation on apoptotic and necrotic cells.13
The lectin pathway shares several components with the classical pathway (see Figure 14-1). Initiation of the lectin pathway is mediated through the binding of mannose-binding lectin (MBL; also known as mannose-binding protein [MBP]) or ficolin to a variety of repetitive carbohydrate moieties such as mannose, N-acetyl-D-glucosamine, and N-acetyl-mannosamine, which are abundantly present on a variety of microorganisms.14 MBL, a plasma protein composed of a collagen-like region and a carbohydrate-binding domain, is structurally similar to C1q. As in the case of the C1qC1rC1s complex, MBL forms complexes in the plasma with other proteins, such as mannose-binding protein–associated serine proteases (MASPs).15 Under physiologic conditions, MBL does not bind to mammalian cells, probably because these cells lack mannose residues on their surfaces. Once bound to microbial pathogens, MASPs undergo conformational changes and become active enzymes that are capable of cleaving C4 into C4a and C4b fragments. At this point, the lectin pathway intersects with the classical pathway, and a C3 convertase, that is, the C4b2a complex, is eventually generated. One study, however, has shown that MBL, in the absence of C4 or C2, is still capable of activating the complement system by engaging the alternative pathway.16 This finding suggests that this unconventional lectin-initiated complement activation process may serve as a “backup” protective mechanism in individuals deficient in C4 or C2.
C3 convertases generated during the first stage of complement activation cleave C3, the central and most abundant component of the complement system. This proteolytic cleavage gives rise to a smaller C3a fragment and a larger C3b fragment. The C3b molecules associate with C4bC2a or with C3bBb complexes to form the classical and alternative C5 convertases, respectively. The C5 convertases cleave C5 to form C5a and C5b. C5b then recruits C6, C7, C8, and multiple molecules of C9, which together form the C5b-9 membrane attack complex (MAC; also called terminal complement complex [TCC]) on the surfaces of foreign pathogens (see Figure 14-1).
Regulators of Complement Activation
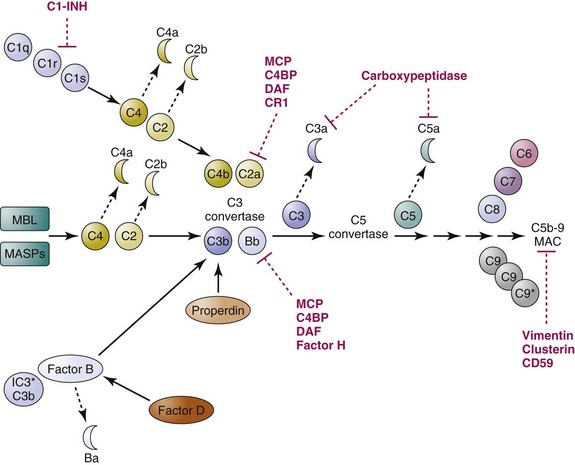
In humans and other mammals, complement activation is controlled by a multitude of regulatory proteins to ensure that this effective machinery is not inappropriately activated on host cells and tissues (see references 17 and 18 for reviews; see Table 14-1). To control the potentially harmful consequence of complement activation, soluble or cell-surface regulatory molecules need to act at multiple steps of the activation pathways using different mechanisms, functioning as proteolytic enzymes (carboxypeptidases, factor I), cofactors for proteolytic enzymes (factor H, complement receptor 1, membrane cofactor protein), protease inhibitors (C1 inhibitor), physical dissociators or competitive inhibitors of multimolecular convertases (C4-binding protein, decay-accelerating factor, factor H), or inhibitors of MAC formation and membrane insertion (CD59, homologous restriction factor). For example, C1 inhibitor (C1-INH) is capable of inactivating C1r, C1s, and MASPs, preventing the activation of the classical pathway and the lectin pathway. Further downstream, plasma carboxypeptidases can quickly remove an arginine residue from the C-terminus of C3a and C5a and generate C3a desArg and C5a desArg, which lose more than 90% of their original biological activities. C3b and C4b are also converted into proteolytic fragments such as iC3b, C3c, C3dg, C4c, and C4d by the serine protease factor I in the presence of cofactors such as factor H and membrane cofactor protein (MCP), thereby preventing the formation of C3 convertases. C3 convertases, once formed on cell surfaces, can be dissembled by the action of decay-accelerating factor (DAF; CD55), C4-binding protein (C4BP), and factor H. Finally, if all these gatekeepers fail, a membrane protein CD59 (also known as “protectin”) will take action to prevent the formation of the MACs, the lytic “terminator” of cells, within the plasma membrane (Figure 14-2).
Receptors for Complement Proteins
Receptors for proteolytic fragments of complement proteins (e.g., C3b, C4b, iC3b, C3d) and, in some circumstances, for complement proteins with altered conformation (e.g., C1q) are expressed by a wide spectrum of cells and serve pivotal roles in executing many of the effector functions of complement previously described (see references 19 and 20 for reviews). Studies have now led to the identification of at least four receptors for C1q, cC1qR (calreticulin; a collectin receptor), gC1qR, C1qRp, and complement receptor 1 (CR1; also know as CD35). Binding of C1q-opsonized immune complexes to endothelial cells via C1q receptors has been reported to induce expression of adhesion molecules on endothelial cells and thus to enhance leukocyte binding/extravasation. On other cell types, C1q binding, presumably via distinct receptors, has been shown to enhance phagocytosis, increase generation of reactive oxygen intermediates, and activate platelets.
Receptors for C3a and C5a have been identified and cloned. The C3a receptor (C3aR) and C5a receptor (C5aR; CD88), which belong to the rhodopsin family of the G protein–coupled 7 transmembrane–domain receptors, are expressed on leukocytes, endothelial cells, podocytes, and proximal tubular epithelial cells in the kidney.21 Interactions of C3aR and C5aR with their respective ligands (C3a and C3a desArg, and C5a and C5a desArg) are essential for the intracellular signaling processes that lead to leukocyte degranulation, production of cytokines, release of vasoactive substances, and other anaphylaxis and inflammatory responses.22
CR1 (CD35) and CR2 (CD21), two major receptors for C3- and C4-derived fragments, belong to the regulators of complement activation (RCA) family.23 CR1 is widely expressed by erythrocytes, neutrophils, monocytes/macrophages, B lymphocytes, some T lymphocytes, and glomerular podocytes. CR1 primarily binds C3b and C4b. One important function of CR1 expressed on erythrocytes is to bind and clear immune complexes. CR1 also plays a role in regulation of complement activation by serving as a cofactor for factor I, which is responsible for cleaving C3b and C4b to iC3b and iC4b, respectively. CR2 is expressed mainly on B lymphocytes, activated T lymphocytes, and follicular dendritic cells, and binds primarily iC3b, C3dg, and C3d. CR2, together with its cognate complement ligands, is a critical link between the innate and adaptive immune systems. For example, coligation of CR2 (as part of the CD19/CD21/CD81 B-cell co-receptor complex) and B-cell receptors on the surfaces of B lymphocytes via the binding of C3d-decorated immune complexes or antigens enhances B-cell activation, proliferation, and antibody production. Antigens and immune complexes opsonized by C3-derived fragments can be retained in the germinal centers of secondary lymphoid follicles via binding to CR2 expressed on follicular dendritic cells; the retained antigens provide essential signals for survival and affinity maturation of B cells as well as for generation of memory B cells.24
Effector Functions of Complement
The complement system is traditionally thought to have the following four biological functions in protecting against invasion by pathogens: (1) initiation of the inflammatory response, (2) opsonization and clearance of pathogens, (3) opsonization and clearance of immune complexes, and (4) osmotic lysis of invading microorganisms.20,25 During SLE pathogenesis, complement activation and its inflammatory consequences are generated by self-antigens and autoimmune complexes rather than by foreign microbes.
The soluble proteolytic fragments, C3a, C4a, and C5a, are highly potent proinflammatory molecules. These anaphylatoxins act as potent chemoattractants to recruit leukocytes into sites of infection or injury and activate these cells by binding to specific receptors (e.g., C3aR and C5aR).22 The larger fragments, C3b, C4b, and their derivatives (e.g., iC3b and iC4b), can remain bound to the surfaces of microbial pathogens (or autoantigens) and facilitate recognition and uptake of the opsonized particles by phagocytic cells. This function is mediated through the binding of these complement-derived fragments to CR1 (for C3b and C4b), CR3 (for iC3b), and CR4 (for iC3b) expressed on phagocytes.
Complement: An Important Bridge between Innate Immunity and Adaptive Immunity
For several decades, the role of the complement system was thought to be limited to the four effector functions already discussed. However, there has been an explosion in discovery of additional roles for complement. A growing number of studies have shown that innate immunity and adaptive immunity, the two arms of the immune system, collaborate in an intricate way to elicit efficient immune responses against infectious agents (see references 24, 26, and 27 for reviews).
The complement system—particularly C3, its derivative fragments, and their cognate receptors—plays an important role in this collaboration. First, C3 plays important roles in B-cell biology. Antigens (and immune complexes) decorated with C3d, the end cleavage product of C3 and a major ligand for CR2, are capable of cross-linking the B-cell receptors to the CR2/CD19/TAPA-1 co-receptor complexes and thus facilitating B-cell activation and enhancing humoral immune responses.24 Second, antigens (and immune complexes) opsonized by C3 can be retained in the germinal centers of secondary lymphoid follicles via binding to CR2-expressing follicular dendritic cells; the retained antigens provide essential signals for survival and affinity maturation of B cells as well as for generation of memory B cells.24 Third, complement is also involved in regulating T-cell activities.26 For example, decay-accelerating factor appears to negatively regulate T cells and prevents T-cell overproliferation during an immune response28; C3a and C5a, via binding to their respective receptors expressed on T cells and antigen-presenting cells, may participate in maintaining T-cell viability, proliferation, and differentiation.29 Fourth, opsonization of pathogens by complement components facilitates their uptake by phagocytes and antigen-presenting cells and thus may enhance presentation of antigens and initiation of specific immune responses. Fifth, complement activation products generated at sites of infection can recruit inflammatory cells and immune effector cells to help eliminate pathogenic antigens. In addition, complement appears to play an important role in opsonizing apoptotic cells and facilitating their clearance (see further discussion later).
Complement and SLE
The involvement of complement in the etiopathogenesis of SLE has been scrutinized over the past several decades. Suffice it to say that the role of complement in SLE is both complex and paradoxically intriguing. On the one hand, activation of the complement system is thought to play an important role in tissue inflammation/damage in SLE as a consequence of tissue deposition of immune complexes formed by autoantigens and autoantibodies.30,31 On the other hand, a hereditary deficiency of a component of the classical pathway (C1, C2, or C4) has been associated with the development of SLE.5 These seemingly discordant roles for complement may be reconciled by studies performed during the past several years. Those studies have demonstrated that, although the complement system plays a role in maintaining immune tolerance to prevent the development of SLE, it also participates in tissue-destructive inflammatory processes once SLE is established in a patient.
Immune Complex Abnormalities, Complement Activation, and Tissue Injury
Considerable evidence has indicated that many of the clinical manifestations and pathology in patients with SLE can be attributed to immune complex abnormalities (e.g., decreased solubility and impaired disposal of immune complexes) and consequent complement activation. In patients with SLE, decreased serum levels of C3 and C4 (due to genetic and/or acquired factors) may not permit sufficient binding of C3 and C4 fragments to the antigen-antibody lattice, thereby preventing the formation of small, soluble immune complexes.32 Furthermore, reduced levels of CR1 on erythrocytes, frequently detected in patients with SLE, may lead to impaired binding, processing, and transporting of immune complexes to phagocytes of the reticuloendothelial system. Consequently, abnormally large quantities of immune complexes are likely to circulate for prolonged periods and form insoluble aggregates that may deposit in various tissues. Alternatively, insoluble immune complexes may form in situ as a result of the “planting” of autoantigens and subsequent binding of autoantibodies at specific loci. Deposited immune complexes do not seem to cause tissue damage directly but provide ample binding sites for complement components. The ensuing activation of the complement system causes the release of various mediators, promotes cellular infiltration and interaction, and culminates in tissue damage. The vascular endothelium and glomerular basement membrane appear to be highly susceptible to this mode of inflammatory damage. This pathogenic sequence provides a molecular basis underlying vasculitis and glomerulonephritis, two hallmark manifestations of SLE.
Complement Deficiency and SLE
Hereditary complement deficiency in humans has been reported for almost every component of the complement system.33,34 Although the overall incidence of hereditary complement deficiency is low in the general population, a deficiency of any complement component is significantly associated with specific human diseases (Table 14-2). The clinical manifestations associated with the hereditary deficiency of individual complement components vary widely and depend on the position of the deficient component within the complement activation cascade.34 Patients with homozygous deficiency of the early components of the classical pathway, C1, C4, and C2, are particularly at risk for development of SLE.5
TABLE 14-2 Complement Component Deficiencies and Associated Diseases
| DEFICIENT COMPONENT | FUNCTIONAL DEFECT(S) | ASSOCIATED DISEASES |
|---|---|---|
| C1 | Impaired clearance of immune complexes and apoptotic cells | Systemic lupus erythematosus (SLE)*,† Glomerulonephritis Bacterial infections |
| C2 | Impaired clearance of immune complexes? | SLE*,† Glomerulonephritis Bacterial infections |
| C4 | Impaired clearance of immune complexes | SLE*,† Glomerulonephritis Scleroderma Sjögren syndrome Bacterial infections |
| C3 | Impaired opsonization; impaired clearance of apoptotic cells? | Bacterial infections*,‡ SLE§ |
| C5 | Impaired chemotaxis; absence of lytic activity | Bacterial infectionsc |
| C6 | Absence of lytic activity | Bacterial infections§ |
| C7 | Absence of lytic activity | Bacterial infections§ |
| C8 | Absence of lytic activity | Bacterial infections§ |
| C9 | Impaired lytic activity | Bacterial infections§ |
| Properdin | Impaired alternative pathway activation | Bacterial infections§ |
| Mannose-binding lectin (MBL) | Impaired lectin pathway activation | Bacterial and viral infections* SLE? |
| C1-inhibitor | Excessive C2 and kininogen activation | Hereditary angioedema* SLE? |
| Factor H | Excessive alternative pathway activation | Atypical hemolytic uremic syndrome (aHUS) Age-related macular degeneration (AMD) Bacterial infections |
| Factor I | Excessive alternative pathway activation | Bacterial infections |
| Membrane cofactor protein (MCP) | Excessive alternative pathway activity | aHUS AMD |
| CD55/CD59 | Excessive MAC formation and cytolysis | Paroxysmal nocturnal hematuria |
† Risk hierarchy for development of SLE: C1 deficiency (~90%) > C4 deficiency (~80%) > C2 deficiency (~30%).
‡ Most frequently infections with encapsulated bacteria, especially Neisseria meningitidis.
§ SLE occasionally reported for patients with homozygous deficiency.
In humans, there are two isotypes of C4, C4A and C4B.35 Complete C4 deficiency (homozygous deficiency of both C4A and C4B) is extremely rare. It was first reported in 1974 in a patient who manifested an acute SLE-like disease,36 and a total of 24 cases have since been reported.5 However, homozygous deficiency of C4A alone and heterozygous deficiency of C4A and/or C4B is relatively frequent. Increased incidences of deficiency of C4A and, less commonly, of C4B, have been reported in patients with SLE.37 A review of clinical cases of SLE associated with complement deficiency revealed a hierarchical correlation between the position of the deficient component within the classical pathway and the prevalence of SLE. It has been estimated that SLE occurs in approximately 90%, 80%, and 30% of patients deficient in C1q, C4, and C2, respectively.5 This risk for SLE in complement-deficient individuals is greater than concordance of this disease in monozygotic twins. This observation indicates that the classical complement pathway loci encode “lupus genes.”
Clinically, patients with SLE associated with homozygous C1q or C1r/s deficiency usually present with symptomatic disease at an early age (before 20 years), have severe disease with prominent cutaneous manifestations, and do not exhibit the usual female predilection.5,34 Similarly, SLE associated with homozygous C4 deficiency often occurs at an early age and manifests cutaneous lesions more frequently than renal disease. Patients with SLE and homozygous C2 deficiency also have less renal involvement but have more cutaneous involvement (especially photosensitivity) and arthralgia. Serologically, the prevalence of antinuclear antibodies (ANAs) and anti–double-stranded DNA (anti-dsDNA) antibodies is often lower in patients with complement deficiency–associated SLE than in patients with idiopathic SLE, but the presence of anti-Ro antibodies appears to be common in SLE associated with C2 or C4 deficiency.
In contrast to the high incidence of SLE and SLE-like disease in patients with homozygous deficiency of the classical pathway components, patients with homozygous C3 deficiency seldom have SLE. Of the reported 24 cases of C3 deficiency, only four patients were described to have SLE-like disease and all tested negative for ANA.5 As for terminal complement components, there have been occasional case reports of patients with SLE and C6 deficiency, C7 deficiency, C8 deficiency, and C9 deficiency. However, the predominant phenotype of homozygous deficiencies of terminal complement components is the recurrence of infection (see Table 14-2).
MBL, the initiating component of the lectin pathway, is structurally homologous and functionally analogous to C1q. Polymorphisms in the promoter and coding regions of the MBL gene that lead to altered serum levels of MBL or encode defective MBL proteins incapable of activating the complement system have been reported. Like C1q deficiency, MBL deficiency has been associated with increased susceptibility to SLE.38 Higher frequencies of an MBL gene variant that encodes a defective MBL protein (due to changes in amino acid residues 54 and 57) have been found in patients with SLE of different ethnic origins. Associations between low serum levels of MBL and SLE have also been reported. Clinically, MBL deficiency in patients with SLE appears to increase their susceptibility to infections.
In patients with SLE, autoantibodies against complement components have also been found to cause acquired complement deficiency. For example, C3 nephritic factor, an autoantibody capable of stabilizing the alternative pathway C3 convertase BbC3b, can cause consumption of complement proteins via unregulated activation of the alternative and terminal pathways.39 Another autoantibody reactive with the first complement component, C1q, has been detected at increased frequencies in patients with SLE. A significant portion of patients with SLE also demonstrate functional C1q deficiency secondary to the presence of anti-C1q antibodies.5 Although the pathophysiologic role of anti-C1q in SLE is largely unknown, its presence in patients with SLE has been associated with lupus nephritis.40






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


