Charcot-Marie-Tooth (CMT) disease is a relatively common inherited familial neuromuscular defect that affects the peripheral nervous system causing weakness, atrophy, and gross slowing of sensory and motor nerve conduction velocities (NCVs). Thus, the condition is classified as a hereditary motor and sensory neuropathy (HMSN). Hereditary peripheral neuropathies, such as CMT, are some of the most common genetic disorders in humans. The disease was named in 1886, when Jean Martin Charcot and his student Pierre Marie in France, as well as Howard Tooth in England, separately described the physical and histologic findings of the same process at about the same time. Charcot’s description of the disease was correct although the presumed pathophysiology of a myelopathy was incorrect. Tooth accurately classified it as a peripheral nerve disorder (
1,
2).
CMT is a demyelinating, hypertrophic neuropathy that produces progressive deterioration of the peripheral nerves, typically causing bilateral, symmetric neurogenic weakness and atrophy. Classically, the disease manifests as distal muscular atrophy of the upper and lower extremities. The distal muscle wasting and weakness usually leads to skeletal deformities. The general pattern of weakness occurs with wasting of the foot and leg muscles that leads to pes cavovarus or equinocavovarus deformity and contractures of the toes. The prolonged selective muscular atrophy caused by the nerve deficit may cause dropfoot, joint contractures, and fixed deformities to develop. Loss of distal proprioception and spinal ataxia are common. The eventual disabling results are an altered steppage gait, shuffling of the feet, and possible confinement to a wheelchair secondary to severe foot deformities. Brewerton et al (
3) reported that two-thirds of all patients who sought treatment for a symptomatic high-arch foot demonstrated an underlying neurologic problem, and half of these patients had CMT disease. In another study, Nagai et al reported that 116/148 (78%) of patients with bilateral cavovarus deformity with no other known existing medical problem who were evaluated by either NCV or CMT DNA Duplication Detection Test (Athena Diagnostics) were diagnosed with CMT. A positive family history of CMT increased the probability to 91% (
4).
Historically, CMT disease was mistakenly termed peroneal muscular atrophy, progressive peroneal atrophy, and peroneal palsy. These misnomers arose because the patient with CMT commonly manifested a cavus foot deformity and apparent weakness of the peroneal musculature. It is now established that the peroneus brevis muscle only appears to be weakened late in the disease process (
2). Weakness of the peroneus longus will be falsely presumed if the primary function of both the peroneus longus and brevis muscles is considered to be eversion of the foot. The peroneus brevis is the primary evertor of the foot. Eventually, the peroneus brevis is typically weakened, and eversion strength is diminished. In addition, eversion weakness is compounded because these muscles are at a functional disadvantage in the cavus foot and thus always appear weak on manual muscle testing.
In contrast, the primary functions of the peroneus longus muscle is to plantarflex and stabilize the first ray against the ground. The peroneus longus is typically one of the last muscles to atrophy and weaken. Even in the cavus foot, the capacity of the peroneus longus to plantarflex the first ray is readily apparent. Thus, the peroneus longus is not one of the first muscles to weaken, but it actually maintains most of its strength until the middle to late stages of the progressive disease process.
In identifying this misconception, Sabir and Lyttle postulated that in CMT disease, muscle degeneration in the lower extremities follows a specific pattern determined as a combination of the following: (a) the muscles supplied by the longest axons of the sciatic nerve are affected first and (b) the muscles with the smallest muscle bulk are the first to show wasting and atrophy (
5). In this manner, the intrinsic muscles of the foot are afflicted initially (contributing to clawing of the digits and pes cavus), followed by the peroneus tertius. Atrophy continues across the anterior leg and affects the extensor digitorum longus, the extensor hallucis longus (EHL), and the tibialis anterior. Occasionally, the EHL tendon is spared, resulting in a hallux hammer toe. Atrophy of the peroneus brevis allows for overpowering of the peroneus longus and tibialis posterior and the exacerbation of the metatarsus primus equinus and cavus deformities. Both the peroneus longus and tibialis posterior muscles are typically preserved inasmuch as they have greater muscle bulk. Supporting this theory, in 17 patients with CMT disease, Fenton et al (
6) found the peroneus longus to be weakened in 42% of the cases and the tibialis posterior to be weakened in only 20% of the cases. In addition, in follow-up of his patients with CMT after surgical correction, Gould (
7) was surprised that the nontransferred peroneal musculature was so strong. He stated, “The power in the peroneal musculature was surprisingly strong since the muscle is no longer under stretch and strain by the previously ‘inverted’ foot especially when a plantigrade status was now achieved.” In a report of 18 patients, he had rated all 18 cases to have poor preoperative peroneal function. After surgery, he found 16 cases with fair peroneal function and 2 cases with good peroneal function.
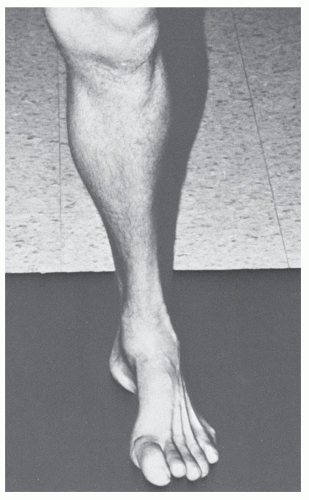
In the most severe cases, the triceps surae may be involved. However, because it has the greatest muscle bulk, it is typically the last muscle group to be affected. Muscles above the knee are generally not affected. The hallmark atrophy in the lower portion of the leg creates a sharp contrast to the normal musculature of the thigh. This appearance has been termed the inverted champagne bottle or stork leg (
Fig. 65.1).
Wasting of the thenar, the hypothenar, and the first dorsal interosseous muscles in the hand is a common finding. Mild clawing of the hand may occur. In more severe instances, an intrinsic muscle deformity of the fingers develops. The appearance of hands demonstrates concavities between the
metacarpals and resembles a monkey fist deformity or skeleton hand (
Fig. 65.2). Sensory loss in the upper limbs, when present, is generally less advanced than in the lower extremities. Hand involvement usually indicates more severe progression of the disease.
Although HMSN was initially described as a single disorder, it is now apparent that they are a heterogeneous group of disorders. The understanding of the histopathology as well as the various types and subtypes of HMSN have increased significantly in the last 15 years.
PATHOLOGY AND MOLECULAR GENETICS
Advances in molecular genetics have greatly changed the understanding of the etiology of this disease. Molecular testing can help establish a secure diagnosis, enable genetic counseling and prognosis, and even aid in the future potential treatment options. The pathogenesis of CMT has been linked to abnormalities/mutations at discrete genetic origins that affect the formation of specific nerve proteins and leads to dysfunction of peripheral nerve axons or their myelin. The major challenge lies in determining the individual contributions by neurons and Schwann cells pathology to help delineate the detailed molecular functions of the proteins associated with CMT (
10).
Mutations in multiple different genes expressed in Schwann cells and neurons cause a variety of overlapping phenotypes. In most instances, the underlying gene defects alter primarily myelinating Schwann cells followed by secondary axonal degeneration. The demyelinating neuropathies therefore develop into functional axonopathies. The abnormalities that that have been identified further define the role of axonal signaling and the molecular architecture of the Schwann cells and neurons in maintaining normal peripheral nervous system function (
11).
The identification of many new alterations/mutations of genes associated with neuropathy demonstrates the role of
axonal transport, abnormal protein trafficking, and the interconnected pathways of Schwann cells and neurons. These pathways include the control of myelin formation and stability, membrane trafficking, intracellular protein sorting and quality control, and may extend to mitochondrial dynamics and basic protein biosynthesis. Identified groups of pathology include myelin protein components, regulators of myelin gene transcription, and intracellular Schwann cell proteins involved in the synthesis, transport, and degradation of myelin components and mitochondrial fusion. Crucial myelin protein components that are affected are peripheral myelin protein 22 (PMP22), protein zero (PO/MPZ), connexin 32 (Cx32/GJB1), and periaxin, which connects myelin to the surrounding basal lamina. Regulators of myelin gene transcription affected are early growth response 2 (EGR2/Krox20, SOX10). Intracellular Schwann cell proteins involved in the synthesis, transport, and degradation of myelin components affected are myotubularinrelated lipid phosphatase (MTMR2) and regulatory binding partner (MTMR13/SBF2, SIMPLE, dynamin 2). Mutations also can affect mitochondrial fusion of factor GDAP1 (
10,
11).
Three genes commonly causing CMT disease encode myelin-related proteins (peripheral myelin protein 22 [
PMP22], myelin protein zero (
MPZ), and connexin 32 [
Cx32]) (
2,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24 and
25,
36). All three genes play an important role in the formation of myelin or maintenance of the peripheral nerves. It is then easy to place CMT into a group of disorders known as
myelinopathies. Demyelinating versus axonal phenotypes is a major consideration in CMT associated with mutations of these genes. Axonal features of diminished amplitudes of compound motor action potentials (CMAPs) and axonal loss eventually lead to length-dependent axonal degeneration. Length-dependent axonal degeneration is likely the basis of distal predominance of the CMT phenotype and neuropathic muscle wasting progressive.
In a study of 205 Japanese patients with CMT, Hattori et al described that the amplitude of CMAPs correlates significantly with distal muscle strength while motor NCV does not, indicating that clinical weakness results from the reduced number of functional large axons, not from demyelination. PMP 22 duplication caused mainly demyelinating phenotypes with slowed motor NCV and demyelinating histopathology. MPZ had two distinct phenotype subgroups with one showing complete demyelination and the other showing complete axonal features. The difference seems to be the nature and position of the mutation. Cx32 mutations showed intermediate slowing of the motor NCV with predominantly axonal features and relatively mild demyelination (
12).
CLASSIFICATION
Genetic testing and electrophysiologic studies are some of the cornerstones in the diagnosis of CMT that have been helpful in defining subtypes of the hereditary neuropathies. Advances in molecular genetics have greatly complicated the classification of these disorders. The rapid advances in molecular genetics and cell biology of hereditary neuropathy have revealed great genetic complexity. Classification of hereditary neuropathies has evolved from a simple clinical to a detailed molecular classification. The molecular classification is not simple to use, and there is no intervention guided by knowledge of any patient’s specific subtype. The logistics of testing for multiple gene mutations are considerable. Proper diagnosis of the CMT subtype may be important for genetic counseling and prognosis but does not help with treatment interventions at this time (
21).
There are two main electrophysiologic groups in CMT corresponding to Schwann cell and axonal pathology. There are those with low conduction velocity categorized as CMT 1 with conduction velocities below 38 m/s. The second group have near-normal conduction velocities of greater than 38 m/s. An intermediate group has NCVs that overlapped the two main groups. The intermediate group has unique disease mechanisms affecting both Schwann cells and axons (
22).
CMT1 is also known as hypertrophic CMT because of the clinically apparent enlarged and firm nerves found in this disorder. The greater auricular nerve and the sural nerve are generally the best nerves to evaluate for this hypertrophy (
8). CMT1 is characterized by repeated demyelination and remyelination that leads to a microscopic abnormality called the onion bulb and enlargement of the nerve that causes slowing of both motor and sensory NCVs.
This condition appears to begin in the feet and legs, with atrophy and myelin remodeling. Further subdivision of CMT1 is based mainly on specific gene mutations or genetic loci by linkage studies. The most frequent form is CMT1A (60% to 90% of all cases of CMT) (
13,
15,
16 and
17,
23). The mode of inheritance is autosomal dominant, and it has been linked to the short arm of chromosome 17. A 1.5-Mb duplication of the 17p11.2 chromosomal region that contains the
PMP22 gene is responsible for the disease secondary to an overexpression of the protein. There is no mutant gene in this instance, but instead, the disease phenotype results from having three copies of a normal gene. Therefore, a gene dosage effect is usually responsible for the myelinopathy (
2). PMP22 is a structural protein of myelin of the peripheral nervous system. PMP22’s most important role is in myelinated Schwann cells. Homozygotes for this locus have a much more severe form of the disease process with an earlier onset of the symptoms as compared with heterozygotes (
18).
A deletion rather than a duplication of the 17p11.2 locus has been linked to a subtype of HMSN, known as hereditary neuropathy with liability for pressure palsy (
13,
24). These patients have clinical evidence of polyneuropathy over potential anatomic sites of nerve entrapment or compression. However, this condition bears no resemblance to CMT1 or CMT2 electrodiagnostically because evidence shows a conduction block and focal versus segmental NCV slowing.
The form
CMT1B (4% to 5% of CMT1 cases) is linked to chromosome 1q22 and is associated with mutations of MPZ, which is a member of the immunoglobulin super gene family and functions as an adhesion molecule helping the compaction of the peripheral nervous system myelin. MPZ is the major protein component of myelin in the peripheral nervous system and acts at the intraperiod and major dense lines as an adhesion molecule keeping the myelin compact in these areas (
13,
19,
25,
26 and
27). Other patients with CMT1 who do not fall into the chromosome 17 or 1 linkage are listed in separate subgroups. The subgroup
CMT1C has been linked to a mutation in the SIMPLE gene on chromosome 16. Another subgroup,
CMT1D, has been linked to a mutation in the early response 2 gene (ERG2 or Krox gene) on chromosome 10q21-q22 (
28).
Type II HMSN (
CMT2) is the neuronal type and differs from type I in that the onset of symptoms occurs later, without nerve hypertrophy, and with either slightly reduced or normal NCVs
that have decreased amplitudes. This is a primarily axonal involvement with neuropathologic evidence of chronic axonal degeneration and regeneration but no evidence of segmental demyelination (
8,
28,
29). The mode of inheritance is primarily autosomal dominant. The distal latency response may be three times normal and may be a valuable adjunct in identifying silent carriers among relatives of the patient. Atrophy and weakness of the small hand muscles are less severe, whereas distal lower extremity weakness is more pronounced (
29). The characteristic stork leg appearance is seen frequently. Because intrinsic hand weakness is less serious and sensory changes are even less marked, these patients retain the ability to perform most of the activities of daily living despite obvious lower extremity atrophy (
30). At this point, no single gene has been identified as a direct association with the axonal CMT2. Research has led to subclassification of CMT2. Alterations in chromosomes 1p35-p36, 7p14, 3q13-q22, 8p21,1q22-q23, and 3q13 have been identified (
13,
28,
31).
The sex-linked variety is the second most common molecularly designated form of HMSN and manifests during the second decade of life and tends to run a more severe course, with marked deformity developing in the third decade. This condition is classified as
CMTX and is linked to mutations in gap junction proteins (GJB1) and connexin 32—the Cx32 gene on chromosome Xq13-q22 (
20,
32). The incidence ranges from 6% of the CMT1 to 20% of all types of CMT. It is characterized by progressive distal muscle atrophy and weakness and areflexia and has variable sensory abnormalities. Males are more commonly and more severely affected. The NCV in these patients varies, with most patients exhibiting decreased values. The gap junction formed by connexin 32 plays an important role in the homeostasis of myelinated axons. Reportedly, mutations of connexin 32 fail to form functional gap junctions. Due to this, there is evidence of demyelinating neuropathy with prominent axonal degeneration. It is inherited as an autosomal dominant disorder 90% of the time, with no possibility of male-to-male transmission. (
13,
20,
32,
33 and
34).
HMSN type III (also termed
Déjérine-Sottas disease) is still used to define severe cases of hypodemyelinating neuropathy of early onset. The criteria for diagnosis are merely based on the severity of the involvement: (a) onset of symptoms by age 2 years; (b) severe motor, sensory, and skeletal deficits with frequent extension to more proximal muscles; (c) NCVs less than 12 m/s; and (d) evidence of severe hypodemyelination on nerve biopsy, with onion bulbs consisting of basal lamina reduplication. Originally, this was thought to be an autosomal recessive inheritance; however, most of the identified mutations occur in the heterozygous form with the genes for
PMP22, MPZ, and
ERG2. Because these are the same genes associated with CMT1, many authors consider CMT1 and Déjérine-Sottas disease simply variants of different severity of the same disease caused by similar mutations (
13).
The most severe disability is found in patients with autosomal recessive inheritance of the disease. Clinical manifestations of the autosomal recessive type of inheritance appear around the age of 8 years, and profound weakness is usually present by the second decade (
34). Therefore, the earlier the onset of the disease, the poorer is the prognosis. Autosomal recessive forms of CMT account for less than 10% of the families but are more common in the Mediterranean basin and in the Middle East. This form is classified as
CMT4 (
13). CMT4 is further subdivided based on linkages to different chromosomes. Autosomal recessive CMT has both demyelinating and axonal forms. Eight genes on chromosome 10q23 and 12p11-q13 (EGR2, GDAP1, KIAA1985, MTMR2, MTMR13, NDRG1, PRX, and CTDP1) have been linked to demyelinating autosomal recessive CMT. Axonal autosomal recessive CMT has been linked to encoding A-type lamins (LMNA), ganglioside-induced differentiation-associated protein 1 (GDAP1), and the mediator of RNA polymerase transcription II (
35). However, because this form of transmission is so rare, further description of these mutations is beyond the scope of this chapter.
Table 65.1 summarizes the genotypes and phenotypes of CMT disease.
INHERITANCE
All modes of genetic inheritance have been reported; however, CMT disease is inherited as a dominant trait in most cases. A family history of thin legs and high arches can frequently be obtained. Skre investigated the inheritance pattern of CMT disease in western Norway (
34). Three methods of transmission were distinguished: (a) autosomal dominant CMT disease, with an estimated prevalence of 36.0 in 100,000; (b) X-linked recessive CMT disease, with an estimated prevalence of 3.6 in 100,000; and (c) autosomal recessive CMT disease, with an estimated prevalence of 1.4 in 100,000.
When the method of transmission is dominant, the disease first manifests at about 30 years of age, tends to progress slowly, and leads to moderate impairment of muscle function. In more than 70% of patients with the autosomal dominant mode of transmission, muscle atrophy is steadily progressive. Less often, the disease arrests completely (23%) or manifests only intermittently (7%) (
34). In autosomal dominant disease, 53% of those affected have mild pes cavus, and 22% have severe cavus deformity (
39). Dyck and Lambert described two types of autosomal dominant CMT disease (
8). Their original subdivision into CMT1 or demyelinating (HMSN type I, hypertrophic neuropathy) and CMT2 or axonal (HMSN type II, neuronal type), based on electrophysiologic and neuropathologic criteria, is still valid (
8,
29). However, the classification scheme of CMT changed with the new understanding of the disease process and how it is related to genetic mutations. Several separate types of HMSN are currently recognized, as compared with Dyck and Lambert’s original classification into hypertrophic
and neuronal forms (
Table 65.1). The previously described newer classifications account for the varying clinical presentation, pathologic conditions, electrodiagnostic criteria, mode of inheritance, and genetic analysis (
13).