Cerebral Palsy
H. Kerr Graham
Pam Thomason
Tom F. Novacheck
INTRODUCTION
Cerebral palsy (CP) is the most common cause of physical disability affecting children in developed countries. The prevalence is about 2 per 1000 live births and is not decreasing. Children with CP have complex needs and are usually managed by a multidisciplinary team. The medical literature dealing with CP is extensive and varies from level I (Randomized Clinical Trials [RCTs]), to cohort studies and case reports (levels IV and V). In this chapter, the most recent and the highest level of evidence will be cited, where possible. However, randomized trials in CP are difficult to perform and relatively few have been published, especially on the orthopaedic aspects of CP. When RCTs are not available, cohort studies, with long-term follow up and objective outcome measures, will be referenced.
CEREBRAL PALSY: DEFINITION
Cerebral palsy was described in 1861 by the English Physician William Little, who recognized a link between difficult births and the development of deformities (1). For many years, CP was known as “Little’s Disease.” Little popularized tenotomy to correct deformity in CP and was the first to bridge the gap between neurology and orthopaedics. Although his understanding of the link between brain injury and deformity has stood the test of time, his views on the causation of CP have been superseded. It is now accepted that only 10% to 20% of CP is related to perinatal events. The influence of “difficult births” may have been much greater in Little’s era, when maternal health was poor, maternal and infant mortality was high, and obstetric services were primitive.
The term cerebral palsy was also used by Sir William Osler in 1889 in a book titled “The Cerebral Palsies of Children” (2). Freud considered CP to be caused not just at parturition but also earlier in pregnancy because of “deeper effects that influenced the development of the foetus” (3). Many other definitions have been proposed and debated since then (4).
THE 2007 REVISED DEFINITION AND CLASSIFICATION OF CEREBRAL PALSY
The revised definition of CP, published in 2007, is as follows:
Cerebral palsy (CP) describes a group of permanent disorders of the development of movement and posture, causing activity limitation, that are attributed to nonprogressive disturbances that occurred in the developing fetal or infant brain. The motor disorders of cerebral palsy are often accompanied by disturbances of sensation, perception, cognition, communication, and behavior, by epilepsy, and by secondary musculoskeletal problems (5).
The new definition has been widely accepted and is recommended as the most useful current operational definition of CP. However, it should be remembered that there is no test, genetic, metabolic, immunologic or otherwise, that demonstrates the existence or absence of CP. There is no specified cause such as cerebral pathology or even type of motor impairment, only that motor impairment exists resulting from nonprogressive cerebral pathology, acquired early in life (6).
BRAIN DEVELOPMENT AND GROSS MOTOR FUNCTION
During the first trimester of pregnancy, the growth of the brain is rapid and the brain differentiates into a recognizable cerebrum, cerebellum, brain stem, and spinal cord at a very early age of fetal development. During this time of explosive growth, the developing brain is highly susceptible to genetic influences, exogenous toxins, nutritional deficiencies and other insults, some of which can be characterized by meconium analysis (7). Neuronal development peaks in the second trimester. Neurons differentiate from neural stem cells around the periventricular regions and migrate centrifugally toward the surface of the cerebral cortex. This results in functional activity in neurons by 7 weeks of differentiation with reflex movements detectable in the fetus by the 15th week of gestation (8). By the end of the second trimester, the majority of neurons have been formed. Loss of neurons can be accommodated by neuronal plasticity but not by the generation of new neurons. The third trimester is characterized by extensive synaptogenesis and remodeling with glialization commencing in the second trimester and continuing at least until the age of 2 years. Myelination of neurons begins late in the third trimester, reaches a peak in the early years of childhood, and continues into adolescence, following a well-defined pattern (9). The myelination of complex pathways results in the progressive elimination of primitive reflexes, during the first 6 months of neonatal life as normal postural reflexes appear and the acquisition of gross motor skills occurs. In the typically developing infant, head control is achieved by age 3 months, independent sitting by 6 months, crawling by 8 months (usually accompanied by pulling to stand) and independent walking by the age of 12 months. However, even typically developing infants may take 3 to 6 months longer than these mean figures and still be considered to have typical development (10).
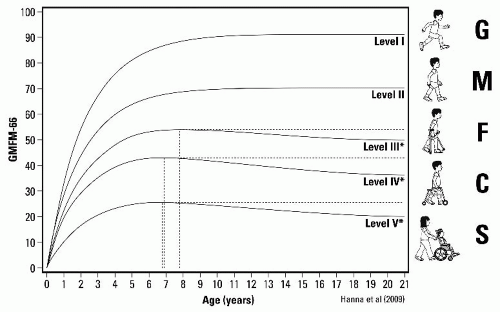 FIGURE 14-1. Gross motor curves in children with CP. The curves are based on longitudinal measurements of gross motor function, using the Gross Motor Function Measure (GMFM). Note the rapid acquisition of gross motor function between birth and age 2 years in all groups. Between the age of 2 and 6 years, the curves reach a plateau and level out into the five levels of the Gross Motor Function Classification System (GMFCS). (From Gallagher C, Sheedy M, Graham HK. Integrated management with botulinum neurotoxin A. In: Panteliadis CP, ed. Cerebral palsy. A multidisciplinary approach. Munchen, Germany: Dustri-Verlag; 2011:213-236, with permission.) |
MOTOR CURVES AND CEREBRAL PALSY
The development of gross motor function in children with CP can be described by a series of curves that were derived from longitudinal measurements of gross motor function, using the Gross Motor Function Measure (GMFM) (11, 12) (Fig. 14-1). The curves show rapid acquisition of gross motor function in infants with a progressive separation of the curves especially between the ages of 2 and 4 years. The curves plateau between the ages of 3 and 6 years. The five gross motor curves constitute the five levels of the Gross Motor Function Classification System (GMFCS) (11, 12, 13, 14 and 15).
Understanding the position of a child’s development in relation to their gross motor curve provides a rational basis for the understanding of management strategies, goal setting, and long-term gross motor function. For example, a 2-year-old child GMFCS level II with signs of spastic diplegia is treated with a physical therapy program, ankle-foot orthoses (AFOs), and injections of Botulinum toxin A (BoNT-A) to the gastrocsoleus and hamstring muscles. Within 3 months, the child is noted to have progressed from standing with support to independent walking. While the intervention may well have contributed to these gains in gross motor function, the child is at the stage of rapid acquisition of gross motor function with or without intervention (12). This underlines the need for intervention studies in the first 6 years of life to be controlled. The popularity of many forms of intervention in early childhood in children with CP is the mistaken attribution of improvements in gross motor function to the intervention, when natural history has an undoubtedly much greater effect. Association is not causation.
In the majority of children aged 6 to 12 years, gross motor function has reached a plateau (Fig. 14-1). At the same time, gait parameters are noted to show deterioration as contractures and bony deformities increase (16, 17, 18 and 19). Changes in gross
motor function and in gait during this plateau can be more realistically attributed to intervention, and longitudinal cohort studies are less liable to misinterpretation than in the birth to 6 years age group.
motor function and in gait during this plateau can be more realistically attributed to intervention, and longitudinal cohort studies are less liable to misinterpretation than in the birth to 6 years age group.
PREVALENCE OF CEREBRAL PALSY AND CAUSAL PATHWAYS
The incidence of CP varies from 1 to 7 children per 1000 live births, according to maternal health, prenatal and perinatal maternal, and child health care services (6, 20). Prevalence rates are accurately reported in countries with well-developed health care services and are most reliable in countries with national CP registers including some European countries and Australia. In these countries, prevalence rates are comparable at around 2 per 1000 live births (20, 21). In most countries, prevalence rates are either static or increasing.
There is a paradoxical relationship between prevalence rates and the provision of neonatal intensive care. Sophisticated neonatal intensive care for premature and low birth weight infants may reduce the risk of brain injury in some and eliminate brain injury in other high-risk neonates. However, the lives of very premature and very low birth weight neonates with a severity of health problems, which would previously have resulted in premature mortality, are saved. These infants may survive with an increased risk of moderate and severe CP (6, 20).
Males are at higher risk of CP, perhaps due to gender-specific neuronal vulnerabilities (22). The risk of CP increases with decreasing gestational age. However, because births before 32 weeks contribute <2% of neonatal survivors, they contribute a minority (20% to 25%) of all CP in developed countries (6, 20, 23). The majority of CP cases are born at term.
The risk of CP increases 4-fold in twins and 18-fold in triplets (24, 25, 26 and 27). The widespread use of in vitro fertilization has greatly increased the rate of multiple births, which has resulted in an increase in CP rates (6, 28). The high CP rates in multiple births are in part explained by shorter gestation and low birth weight, but these are not the only factors. Any factor causing preterm birth may lie on a causal pathway to CP (6).
CENTRAL NERVOUS SYSTEM PATHOLOGY AND ETIOLOGY
CP is the most common cause of the upper motor neuron (UMN) syndrome in childhood, a syndrome characterized by positive features (spasticity, hyperreflexia, and co-contraction) and negative features (weakness, loss of selective motor control, sensory deficits, and poor balance) (Fig. 14-2). Clinicians have traditionally focused more on the positive features because it is possible to treat spasticity. However, it is the negative features, which determine the locomotor prognosis. Weakness and loss of selective motor control determine when or if a child will walk. Balance deficits may dictate long-term dependence on a walking aid (13, 15).
The brain lesion, which results in CP, is a “static encephalopathy.” In other words, the brain lesion is not progressive and is unchanging. This is in contrast to musculoskeletal pathology in the limbs, which is progressive and constantly changing during growth and development (6, 29, 30).
At least 70% of cases have antecedents during pregnancy and only 10% to 20% have any relation to the child’s delivery (6, 20, 31). The mechanism of causal pathways suggests that in any one case of established CP a number of factors may have contributed to the brain lesion resulting in the specific clinical phenotype. There are a number of genetic predispositions to CP that may require an addition of an environmental trigger such as a maternal infection to be expressed as a brain lesion and CP. The genotype may load the gun and the environment pulls the trigger. A large number of major brain malformations have a genetic basis, and subtle genetic polymorphisms may also play a role (31, 32).
About 10% of infants with CP weigh <1500 g at birth. In this low birth weight group, the risk of CP is 90 per 1000, compared to 3 per 1000 in infants born at term and weighing more than 2500 g. Maternal risk factors include viral infections, urinary tract infections in late pregnancy, dietary deficiency, some prescription drugs, drug or alcohol abuse, maternal epilepsy, mental retardation, hypothyroidism, preeclamptic toxemia, cervical incompetence, and third-trimester bleeding. Obstetric risk factors include multiple births, placental abruption, premature rupture of membranes, chorioamnionitis, and prolonged labor. Other obstetric factors include the administration of oxytocin, cord prolapse, and breech presentation, when accompanied by low Apgar scores (6, 28). Neither routine use of fetal monitoring during labor nor increasing rate of Cesarean section has resulted in a decrease in the prevalence of CP (6).
The older the child at the time of the acquired brain lesion, the more the clinical syndrome is likely to differ from classical CP. Age 2 to 3 years is an important watershed (5).
CP was formerly a clinical diagnosis with occasional confirmation by central nervous system (CNS) imaging. With the availability of MRI and safer anesthesia for children, the majority of children suspected of having a CP will have brain imaging (31). A recent practice parameter from the American Neurological Association recommended that the diagnosis of CP be confirmed by imaging (33). In a large recently published multicenter study, the brain lesions identified by MRI are given in Table 14.1.
On the basis of their MRI scans, only 20% of cases of CP were considered as possibly being secondary to some type
of obstetric mishap. Twelve percent of cases, with a clinical diagnosis of CP, had a normal MRI scan (31).
of obstetric mishap. Twelve percent of cases, with a clinical diagnosis of CP, had a normal MRI scan (31).
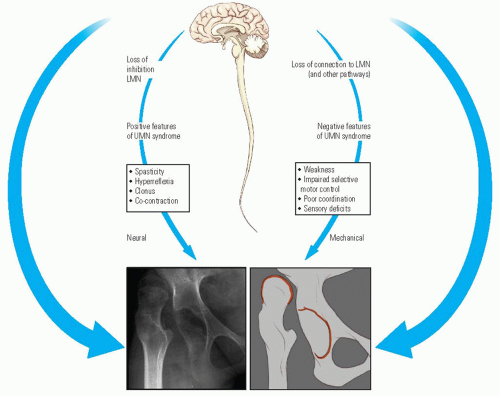 FIGURE 14-2. CP is a neuromusculoskeletal disorder. The CNS lesion has profound effects on the growing skeleton leading to deformities in both the upper and the lower limbs. Note the effects of both the positive and the negative features of the Upper Motor Neuron (UMN) syndrome. |
PROGRESSIVE MUSCULOSKELETAL PATHOLOGY: CP IS A NEUROMUSCULOSKELETAL DISORDER
By definition CP is a static encephalopathy, but the musculoskeletal pathology is progressive (6, 30). Chronic neurologic impairment affects the development of bones and muscles (Fig. 14-3). In spastic hemiplegia, the affected side demonstrates muscle atrophy and limb shortening, compared to the unaffected side. Thus, CP is a neuromusculoskeletal disorder (29, 30).
The key feature of the musculoskeletal pathology in CP is failure of longitudinal growth of skeletal muscle. An apt synonym for CP is “short muscle disease.” The conditions for normal muscle growth are regular stretching of relaxed muscle, under physiologic loading conditions and normal levels of activity (29). In children with CP, skeletal muscle does not relax during activity because of spasticity and the
children have greatly reduced levels of activity because of weakness and poor balance (30). In animal models of CP, such as the hereditary spastic mouse, there is failure of longitudinal muscle growth, in relation to bone growth. Affected mice develop equinus deformities because of a failure of longitudinal gastrocsoleus muscle growth when compared to tibial growth (34). However, muscle growth can be enhanced by the injection of BoNT-A soon after birth (35). The newborn child with CP does not have contractures or lower limb deformities, and most do not show signs of spasticity (29, 30). With time, spasticity develops, activity levels remain low, the growth of muscle-tendon units (MTUs) lags behind bone growth and contractures develop. Although the MTU is short in CP, muscle fibers may not be short (36, 37). Because of disordered growth and abnormal biomechanics, torsional abnormalities persist or develop in the long bones and instability of joints including the hip and subtalar joint develop. Eventually, premature degenerative arthritis may develop (6, 30, 38, 39, 40 and 41).
children have greatly reduced levels of activity because of weakness and poor balance (30). In animal models of CP, such as the hereditary spastic mouse, there is failure of longitudinal muscle growth, in relation to bone growth. Affected mice develop equinus deformities because of a failure of longitudinal gastrocsoleus muscle growth when compared to tibial growth (34). However, muscle growth can be enhanced by the injection of BoNT-A soon after birth (35). The newborn child with CP does not have contractures or lower limb deformities, and most do not show signs of spasticity (29, 30). With time, spasticity develops, activity levels remain low, the growth of muscle-tendon units (MTUs) lags behind bone growth and contractures develop. Although the MTU is short in CP, muscle fibers may not be short (36, 37). Because of disordered growth and abnormal biomechanics, torsional abnormalities persist or develop in the long bones and instability of joints including the hip and subtalar joint develop. Eventually, premature degenerative arthritis may develop (6, 30, 38, 39, 40 and 41).
TABLE 14.1 Frequency of Brain Pathology by MRI | ||||||||
|---|---|---|---|---|---|---|---|---|
|
An important therapeutic window exists for spasticity management before the development of fixed contractures (38, 39) (Fig. 14-3). A second therapeutic window exists for the correction of fixed musculoskeletal deformities, before the onset of decompensation (39). There are three important longitudinal studies of gait in children with spastic diplegia that confirm that the musculoskeletal pathology and the attendant gait disorder are progressive during childhood. These studies provide an important insight into natural history and a framework to interpret the results of surgical intervention (16, 17 and 18).
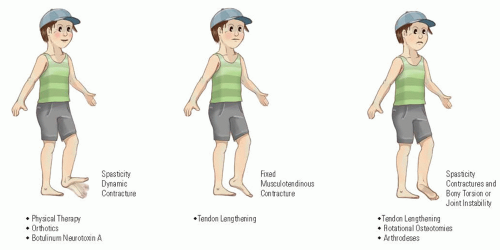 FIGURE 14-3. Musculoskeletal pathology in children with CP is progressive. At Stage 1, children have spasticity but no fixed contractures and can be managed nonoperatively. At Stage 2, there are fixed contractures and at Stage 3 contractures and bony deformities that may require corrective orthopaedic surgery. (Modified after Dr. Mercer Rang.) |
CLASSIFYING CEREBRAL PALSY
CP may be classified by the cause (when known) and the brain lesion as determined by MRI. Classification by movement disorder, topographical distribution, and gross motor function may be relevant to management, including orthopaedic surgery (13, 31, 42, 43) (Figs. 14-4 and 14-5A,B).
Movement Disorder.
The most common approach to classification by movement disorder divides the disorders into pyramidal (spastic) and extrapyramidal (dystonic, athetoid) types (44). The majority of children with CP show features of both pyramidal and extrapyramidal involvement. When there is involvement of both pyramidal and extrapyramidal systems, spasticity and dystonia may coexist to varying degrees (43).
Spasticity.
Spastic CP is by far the most common subtype and in most series comprises between 60% and 85% of all cases (21, 29, 43, 44, 45 and 46) (Fig. 14-4). A large population-based study of children with CP found that 85% of children had a primarily spastic movement disorder (21). Classically, spasticity is the result of a lesion affecting the pyramidal system and results in velocity-dependent increase in muscle tone with increased spastic tonic stretch reflexes. Spasticity is often associated with prematurity and the characteristic lesion of periventricular leucomalacia (PVL) on MRI (31).
Dystonia.
Dystonia is the second most common form of movement disorder in CP and may not develop until late
childhood. Dystonia is often underreported in CP registers and population-based reviews (21, 45). Dystonia is diagnosed by observing abnormal twisting postures and writhing movements that vary in intensity. Dystonia may be triggered or worsened by attention, distraction, startling, overuse, fatigue, touch, or pain (43). Resting tone is variable and postures in both the upper and lower limbs vary with time. The brain lesion resulting in dystonia is usually in the basal ganglia and is more likely to be associated with a child who has had a term birth and widespread white matter lesions (31, 46). Uncontrolled movements seen in response to stimulation of the nervous system (e.g., volitional movements, loud noises, pain, etc.) are considered dystonic, while similar uncontrolled movements seen at rest are athetoid (43). Oral medications such as L-dopa and Artane may be beneficial in some children with dystonia. Severe dystonia can be managed by intrathecal baclofen (ITB) pump (38, 46) (Fig. 14-5A,B).
childhood. Dystonia is often underreported in CP registers and population-based reviews (21, 45). Dystonia is diagnosed by observing abnormal twisting postures and writhing movements that vary in intensity. Dystonia may be triggered or worsened by attention, distraction, startling, overuse, fatigue, touch, or pain (43). Resting tone is variable and postures in both the upper and lower limbs vary with time. The brain lesion resulting in dystonia is usually in the basal ganglia and is more likely to be associated with a child who has had a term birth and widespread white matter lesions (31, 46). Uncontrolled movements seen in response to stimulation of the nervous system (e.g., volitional movements, loud noises, pain, etc.) are considered dystonic, while similar uncontrolled movements seen at rest are athetoid (43). Oral medications such as L-dopa and Artane may be beneficial in some children with dystonia. Severe dystonia can be managed by intrathecal baclofen (ITB) pump (38, 46) (Fig. 14-5A,B).
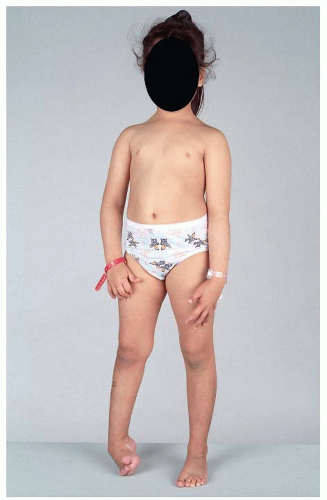 FIGURE 14-4. This girl has a right spastic hemiplegia. Her upper limb involvement is “dynamic.” She has spasticity in her elbow flexors and this becomes apparent when she walks and even more pronounced when she runs. She has a severe varus deformity affecting her right foot because of spasticity in both tibialis anterior and tibialis posterior. The varus posture is consistent and does not change from day to day. Spastic CP tends to be predictable and amenable to corrective orthopaedic surgery. |
Mixed Movement Disorder.
Many children with CP born at term have extensive brain lesions on MRI and have a mixture of pyramidal and extrapyramidal movement disorders. Defining the major movement disorder and the secondary and associated disorders can be challenging (43, 46). Dystonia and spasticity may occur in the same limb segments and distinction requires separation of the velocitydependent from the action-induced and posture-responsive components of the hypertonia. Spasticity is evaluated by passively moving limb segments and joints at variable speeds. Dystonia is more easily confirmed by observation than palpation (39).
Ataxia.
Ataxia may be part of a genetically determined syndrome and is uncommon as a pure movement disorder in CP (21, 45). Ataxia is disturbance of coordination and therefore most easily observed during walking. There may be associated signs of cerebellar dysfunction, including tremor. Because the resting tone is normal, and the majority of children have quite good walking ability, contractures are uncommon, and children with pure ataxia do not develop hip dysplasia or scoliosis (30).
Hypotonia.
Many children who develop hypertonic CP are initially hypotonic, and this phase may last for several years. Hypertonia may not be expressed until myelination reaches a certain stage of completion (9) Many children with intellectual disabilities and certain other syndromes exhibit hypotonia, joint laxity, and developmental delay. Unless there is, in addition to the hypotonia, a defined static brain lesion, these children are not classified as having CP (6, 30).
Topographical Distribution
Unilateral Bilateral
1. Unilateral
Monoplegia, hemiplegia
2. Bilateral
Diplegia, quadraplegia
Topographical distribution is a classification of CP according to which limb segments are affected. As with classification by movement disorder, there is considerable variability in the terminology used, especially between different countries. The emphasis in Europe is the subdivision of CP into unilateral and bilateral types (44). The unilateral types can be subdivided into monoplegia (affecting only one limb) and hemiplegia (affecting one side of the body. The majority of children who seem to have a lower limb monoplegia with spastic equinus and toe walking, when asked to run, will show some abnormal upper limb posturing (21).
The common forms of bilateral CP are diplegia and quadriplegia (21, 45). Children with diplegia have bilateral lower limb involvement that may be symmetric or asymmetric. The involvement of the upper limbs is restricted to deficits in fine motor function, and overall upper limb function
is good. Spastic diplegia is most commonly associated with prematurity and PVL (31). The affected children usually have normal intelligence and a good prognosis for independent walking although many have visual deficits and learning difficulties. Quadriplegia refers to involvement of the upper and the lower limbs, and such children are usually born at term and have extensive brain involvement (46). They usually have a mixed movement disorder with spastic and dystonic features. The severity of involvement varies between the upper and the lower limbs and between the two sides. Because of the greater degree of brain involvement, quadriplegia is far more likely to be associated with comorbidities such as seizure disorder, learning challenges, and impairments of speech or cognition (5).
is good. Spastic diplegia is most commonly associated with prematurity and PVL (31). The affected children usually have normal intelligence and a good prognosis for independent walking although many have visual deficits and learning difficulties. Quadriplegia refers to involvement of the upper and the lower limbs, and such children are usually born at term and have extensive brain involvement (46). They usually have a mixed movement disorder with spastic and dystonic features. The severity of involvement varies between the upper and the lower limbs and between the two sides. Because of the greater degree of brain involvement, quadriplegia is far more likely to be associated with comorbidities such as seizure disorder, learning challenges, and impairments of speech or cognition (5).
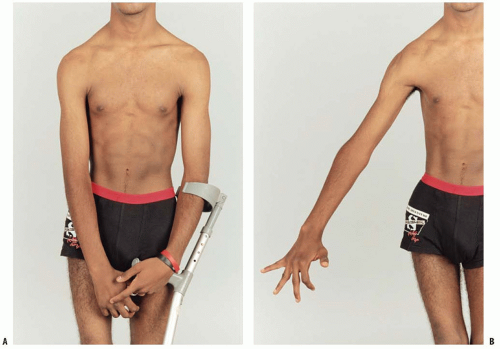 FIGURE 14-5. A and B: This teenage boy also has a right hemiplegia but his movement disorder is dystonic. He is using his uninvolved left hand to restrain his right hand. When the restraint is removed, his right arm undergoes variable dystonic posturing, with abduction at the shoulder, extension at the elbow, and abduction of the fingers. His dystonia is so severe that he requires a crutch and functions at GMFCS level III. Dystonic posturing can rarely be improved by orthopaedic surgery but may respond to medications including ITB. |
A small number of children appear to have a “triplegia” or three-limb involvement. This usually is a combination of hemiplegic involvement with both lower limbs involved to an asymmetric degree. One upper limb seems to be largely spared. This is an uncommon type of CP and is sometimes grouped with spastic quadriplegia (42, 45). Classification by topographical distribution is not a strictly functional classification, but there are functional implications. Almost all children with hemiplegia walk independently in the community, about 80% of children with diplegia walk either independently or with assistive devices, but only 20% of children with quadriplegia walk and then only with assistance (21). The separation of children into diplegia and quadriplegia is arbitrary and unsatisfactory, hence the need for a classification based on a valid and reliable measure of gross motor function (13, 42, 44).
Classification by Gross Motor Function.
The development of the GMFCS has for the first time given a common language to communicate about CP (13). The GMFCS is a five-level ordinal grading system in which a series of descriptors, supplemented by illustrations, can be used in five different age groups to classify gross motor function in CP (Figs. 14-6 and 14-7). The GMFCS has established validity (based on the GMFM), reliability, and stability. There is good agreement between clinicians and also between clinicians and parents (47). Given that the GMFCS is a grading system and not an outcome measure, it is the major prognostic information which must be considered in all children with CP. Knowing a child’s long-term gross motor prognosis has management implications. For example, a child between the ages of 6 and 12 years at GMFCS level IV may perform some stepping with a heavily adapted walker, under the supervision of a therapist or a parent (13). However, following the pubertal growth spurt, useful walking is not sustained (15). It would be inappropriate to offer such children invasive treatments to improve or prolong walking because these will not be successful in the long term. Appropriate goal setting would be maintaining standing transfers (15).
 FIGURE 14-6. GMFCS E & R (Expanded and Revised) between 6th and 12th birthday: Descriptors and illustrations. |
 FIGURE 14-7. GMFCS E & R (Expanded and Revised) between 12th and 18th birthday: Descriptors and illustrations. |
The prevalence and severity of medical comorbidities shows good correlation with GMFCS (48). Severe respiratory disease, nutritional deficiencies, and premature mortality are largely seen at GMFCS levels IV and V. Children who are at GMFCS levels I and II do not have severe medical comorbidities (apart from epilepsy) nor do they show significant excess mortality.
Certain musculoskeletal features and deformities are also closely related to GMFCS level. The shape of the proximal femur shows a strong correlation with GMFCS level. Femoral neck anteversion (FNA) increases from GMFCS level I to level III and then plateaus at a mean of 40 degrees at GMFCS levels III, IV, and V. Mean neck shaft angle (NSA) increases stepwise from GMFCS levels I through to V (49). The incidence and severity of hip displacement is directly predicted by GMFCS level. In one study, children at GMFCS level I showed no hip displacement and those at GMFCS level V had a 90% incidence of having a migration percentage in excess of 30% (50). The relationship between GMFCS and hip displacement has direct implication for screening and management protocols.
CEREBRAL PALSY: COMORBIDITIES
The spinal cord is not involved in CP and the most common neurologic impairment is epilepsy, affecting about 30% of children. Epilepsy is most commonly seen in hemiplegia, especially when it is acquired postnatally and in children with quadriplegia (48, 51). Intellectual disability is variable and more severe in children at GMFCS levels IV and V. However, many children at GMFCS levels I to III exhibit learning difficulties, autism spectrum disorders, and behavioral and emotional difficulties. Impairments of hearing, speech, and vision are also common and may adversely impact schooling and learning. The prevalence of visual problems is so high (about 50%) that routine screening is advised (52).
Respiratory Disorders.
Children with CP have excess mortality at GMFCS levels IV and V, the levels previously described as spastic quadriplegia. The commonest cause of death is from respiratory disease especially aspiration pneumonia (48, 53). A major risk factor for respiratory disease is “pseudobulbar palsy” in which there are varying combinations of impaired swallowing, esophageal reflux, aspiration, and chest infection. Nocturnal coughing and asthma are also very common. The management of chronic respiratory disease involves careful assessment of swallowing and may necessitate investigation of the upper gastrointestinal tract. Fundoplication and the use of feeding tubes are often beneficial in the management of severe respiratory disease, when aspiration has been confirmed to be a major contributory factor (54).
Gastrointestinal System.
The most severe problems are in GMFCS levels IV and V (48). Pseudobulbar palsy leads to impaired swallowing, vomiting, esophageal reflux, and aspiration. Oral feeding can be slow and inefficient leading to chronic malnutrition, impaired growth, and poor nutritional reserves (55). Malnourished children are at greatly increased risk of postoperative complications after hip reconstruction and scoliosis surgery. Assessment of nutritional status includes a careful dietary history and a radiologic assessment of swallowing in those with suspected gastroesophageal reflux. Nutritional status can be assessed by measurement of serum albumin, iron, and transferrin levels and examination for iron deficiency anemia. Correction of malnutrition may require the introduction of improved oral feeding. In many children, a percutaneous enterogastrostomy or a jejunostomy may transform the child’s nutritional status and general health. Gastroesophageal reflux may respond to medical management but often requires a fundoplication (55, 56).
Of all problems affecting the gastrointestinal system, slow transit and chronic constipation is by far the most common (56). The excessively loaded colon is often noted as an incidental finding during hip surveillance radiographs. Chronic constipation assumes even more importance in the perioperative period when fecal impaction, vomiting, and the inability to achieve satisfactory intake of food and fluids postoperatively causes severe distress and results in prolonged hospitalization. Strategies to ensure that children at GMFCS levels IV and V have a satisfactory bowel management program prior to admission for orthopaedic surgery are advised.
DIAGNOSIS AND ASSESSMENT: THE DIAGNOSTIC MATRIX
In the majority of children who are referred to an orthopaedic surgeon, the diagnosis of CP has already been established by a pediatrician or a pediatric neurologist. The exception is a small number of children with mild hemiplegia and monoplegia who present after walking age with toe walking or some other subtle disturbance of gait. The postural abnormalities associated with monoplegia and hemiplegia are best observed by asking the child to walk and then run in a sufficiently long, well-lit corridor (Fig. 14-8).
History.
The “neonate at risk” and the majority of children with mild CP are diagnosed because they are observed to have
a delay in reaching gross motor milestones. A knowledge of the normal age (and range) of achieving these milestones is important. In typically developing infants, head control is acquired at age 3 months, sitting at 6 months, crawling and pulling to stand at 8 to 9 months, and independent walking at 12 months (10).
a delay in reaching gross motor milestones. A knowledge of the normal age (and range) of achieving these milestones is important. In typically developing infants, head control is acquired at age 3 months, sitting at 6 months, crawling and pulling to stand at 8 to 9 months, and independent walking at 12 months (10).
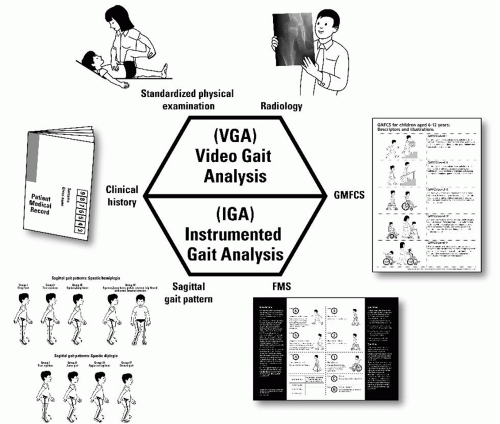 FIGURE 14-8. The diagnostic matrix consists of a standardized approach to clinical history, physical examination, radiology, GMFCS, functional scales (FMS and FAQ), and sagittal gait pattern identification. These components can be used in conjunction with either video gait analysis (VGA) or instrumented gait analysis (IGA). (Illustration copyright © Kerr Graham, Bill Reid and Adrienne Harvey.) |
Recognition of abnormal muscle tone is also important (43). This may include hypotonia in the infant and hypertonia in the child. The mothers of children with CP, who have previously had a typically developing child, often sense that “something is wrong” at a very early stage. It is clearly wise to take the concerns of an experienced parent seriously. Details of the history of the pregnancy, the birth, and early development are very important in establishing the diagnosis of CP. The family history is important to detect such conditions as hereditary spastic paraplegia (HSP) and congenital ataxias (57).
Physical Examination.
Physical examination is important in both preliminary examinations to establish the diagnosis of CP and in subsequent assessments in which the child’s tone, gross motor function, and secondary musculoskeletal pathology are evaluated:
Observation of posture, movement and gait
Assessment of gross motor function: GMFCS, FMS, and FAQ
Evaluation of muscle tone by a combination of observation, palpation, and testing of reflexes.
Assessment of soft-tissue contracture by evaluation of passive joint range of motion and muscle length measurement.
Assessment of torsional abnormalities in the long bones: FNA; tibial torsion; deformities of the spine, hands, and feet.
Sensory evaluation: especially the hemiplegic upper limb.
Assessment of Gross Motor Function: GMFCS.
Knowing the child’s GMFCS level is fundamental in establishing gross motor prognosis and monitoring changes. The GMFCS can be easily and reliably determined by age 4 years and all orthopaedic surgeons should be competent in assigning a grade after the age of 6 years. Changes in GMFCS levels should be carefully documented. The most common reason for a change in GMFCS is an error in the previous or current examination (58). Given that the GMFCS is a categorical grading system, true changes in GMFCS level sometimes occur and these may occur in both directions, that is improvement or deterioration. After major intervention such as selective dorsal rhizotomy (SDR) or multilevel surgery, a small number of children move up a level, but this is uncommon and should not be expected in more than about 5% to 10% of a study population. Deterioration in GMFCS level is more common. Lengthening of the Achilles tendons in children at GMFCS level II can result in progressive crouch gait and the need for assistive devices. Such children may deteriorate from GMFCS level II to III (59).
The GMFCS is a classification system and not an outcome measure. It is expected to be stable throughout a child’s growth and development but is often misused as a proxy outcome measure in intervention studies. Simple scales of gross motor function that can be used as outcome measures are the Functional Mobility Scale (FMS) and the Functional Assessment Questionnaire (FAQ) (60, 61).
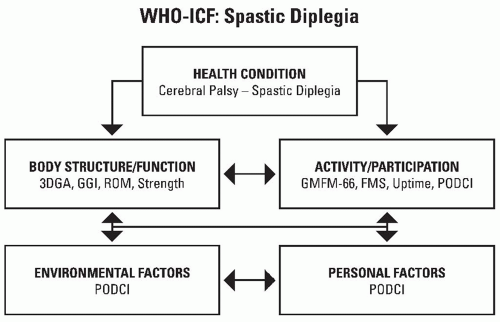 FIGURE 14-9. The World Health Organization International Classification of Functioning (WHO-ICF) as applied to spastic diplegia. Suggested assessment tools are indicated in each domain. (3DGA, three-dimensional gait analysis; GGI, Gillette Gait Index; PODCI, Pediatric Outcomes Data Collection Instrument.) |
The World Health Organization’s International Classification of Functioning (ICF) describes health conditions in several domains, including body structure and function, activities, and participation (62) (Fig. 14-9). These domains are modified by environmental factors and personal factors. Various tools exist to measure parameters relevant to CP in the ICF domains and new measurement tools are being developed. Fortunately, there are now valid and reliable tools to classify and measure gross motor function in both the upper and lower limbs.
Functional Mobility Scale.
The Functional Mobility Scale describes the level of assistance a child requires to mobilize in three different environments, the home (distances of up to 5 m), school (distances of up to 50 m), and the community (distances of 500 m) (60). Hence, three numbers are assigned depending on the level of assistance required in each of these settings. For example, a child at GMFCS level III is frequently capable of independent walking in a sheltered familiar environment such as the home. The same child may require the use of Canadian crutches to move around in the school environment but may be too slow to keep up with the rest of the
family during trips to the shopping mall, when a wheelchair may be preferred. The FMS grading for such a child is 5, 3, 1. Unlike the GMFCS, the FMS was designed as an outcome measure and is sensitive to change (59, 63). For example, children at GMFCS level III often require a posterior walker to ambulate prior to multilevel surgery. After optimum biomechanical realignment and correction of spastic contractures, these children can often progress to lesser levels of support. Some will be able to walk increasing distances independently; others will require crutches or sticks when previously they were dependent on a posterior walker. These important changes can be monitored and reported using the FMS (63).
family during trips to the shopping mall, when a wheelchair may be preferred. The FMS grading for such a child is 5, 3, 1. Unlike the GMFCS, the FMS was designed as an outcome measure and is sensitive to change (59, 63). For example, children at GMFCS level III often require a posterior walker to ambulate prior to multilevel surgery. After optimum biomechanical realignment and correction of spastic contractures, these children can often progress to lesser levels of support. Some will be able to walk increasing distances independently; others will require crutches or sticks when previously they were dependent on a posterior walker. These important changes can be monitored and reported using the FMS (63).
Functional Assessment Questionnaire.
The Gillette FAQ is a 10-level, parent report walking scale that describes a range of walking abilities across the spectrum of CP, from nonambulatory to independent ambulation at a high level. In addition to the 10-level walking scale, there is an additional list of 22 items describing a variety of higher-level functional activities requiring varying degrees of walking ability, balance, strength, and coordination. The scale has been shown to be reliable and validity has been established by comparison with other scales used in neuromuscular diseases. It has also been shown to have sensitivity to change with improvements seen in children with CP after such interventions as SDR and singleevent multilevel surgery (SEMLS). It is a simple scale, which can be quickly completed by parents or caregivers and provides an excellent longitudinal view of the child’s gross motor and walking abilities.
Additional Diagnostic Tests.
Longitudinal Assessments with Radiology: Hip Surveillance.
The early stage of hip displacement is silent and formal screening by radiographs of the hips with careful positioning is advised (64, 65). The frequency of such radiographs should be directly related to the risk of hip displacement which is in turn related to the child’s GMFCS level. At GMFCS level I, there is little risk of hip displacement and radiographs are only required if there are findings on clinical examination. More regular radiographs are required at GMFCS levels II and III. Radiographs every 6 to 12 months may be required at GMFCS levels IV and V to monitor progressive hip displacement (66).
Deformities of the feet are very common in all GMFCS levels. Standing weight-bearing radiographs of the feet are extremely useful in the longitudinal assessment of progressive foot deformity. Recently, a series of radiologic indices have been published in typically developing children that are an excellent baseline with which the results in CP can be compared (67).
Instrumented Gait Analysis.
Evaluation of gait and functioning in children with CP can be considered in the format of a diagnostic matrix (68). The role of instrumented gait analysis (IGA) is crucial to the evaluation of gait dysfunction, especially in relation to planning and assessing the outcome of major interventions such as SDR and SEMLS (69). Historically, problems with reliability undermined the utility of IGA and the confidence in using such information for planning interventions (70, 71 and 72). Recent work from several centers has reestablished confidence in the reliability of gait kinematics (73, 74). Only about half of the orthopaedic surgeons in North America who care for children with CP have access to IGA (75). There are children with symmetric gait deviations that through the eyes of experienced examiners are relatively easy to recognize. Even for those with experience, there are many children with complex movement disorders, asymmetric gait patterns, and complexities that require IGA for understanding and planning (40, 69). As gait analysis becomes more reliable, cheaper, and more accessible, the quality of assessments and outcomes should continue to improve.
Video Gait Analysis.
Even in centers with access to IGA, video gait analysis (VGA) is a central part of the diagnostic matrix (38). A visual record of a child’s gait and functioning on digital video is of much greater value than observational gait analysis and a written report (39). Digital video can be archived in a permanent fashion, is objective, and can be shared by multiple observers over time. It allows observation and recording of gait from multiple viewpoints, can be replayed in slow motion, and can be reviewed repeatedly, including late in the evening prior to an operating list, when real-time observation of a child’s gait is not feasible (40). In an effort to quantify and objectify the outcome of observational gait analysis, a number of gait scores have been developed of varying degrees of complexity, sophistication, and reliability. These include the Physician Rating Scale, the Observational Gait Scale, and the Edinburgh Visual Gait Score (76, 77 and 78).
VGA has wider application than IGA. IGA requires a child to be about a meter tall and to be able to follow simple commands over a 2-hour testing period. There is much useful information to be gained in children with hemiplegia and diplegia from when they first start to stand and walk which cannot be obtained from IGA. Longitudinal assessment of children following interventions such as injection of BoNT-A, the prescription or modification of orthoses, is also conveniently achieved using serial VGA (39, 79). Gait deviations for children at GMFCS IV may be so severe that the extra cost and effort of IGA may not be necessary (15). VGA and physical exam may be all that is needed.
PHYSIOTHERAPY AND OCCUPATIONAL THERAPY IN CP
Physiotherapy is the most popular and widely used management strategy in children with CP (80, 81, 82 and 83). Some have access to therapy services integrated within the school program and others outside of the school program. The frequency of such programs may be related to the severity of the individual child’s involvement but rarely reflect the family and the child’s real needs. This type of background physical therapy support is valued by the parents of children with CP. These programs provide education and emotional support to the parents coping with the diagnosis of CP and starting the journey through the uncharted waters of raising a child with a lifelong physical disability. Roles that may be assumed by therapists may include education, counseling, coordination of access to other services including physical medicine and rehabilitation, orthotics, and orthopaedic surgery. The parents will often seek the view of the child’s therapist on recommendations for spasticity management, the type of orthosis, and the timing and type of surgical intervention (80). The need for clear communication and teamwork within the multidisciplinary team is obvious. These “background” programs of physical therapy are rarely adequate to ensure an optimum rehabilitation from major interventions such as SDR or SEMLS. Therapy around such episodes needs to be carefully planned and is often best as a team approach involving the child’s community therapist as well as providing the additional therapy in the tertiary hospital or rehabilitation center.
The physical therapy management of children with CP has been based on a variety of theoretical perspectives. The theoretical frameworks most commonly applied can be broadly categorized into biomechanical (splints, orthoses, stretching, and strength training), neurodevelopmental, cognitive (including conductive education [CE] and motor learning), and constraint-induced movement therapy (CIMT) (80, 81, 82 and 83). These approaches are not mutually exclusive. They can have shared components and most therapists use a combination of approaches. Given that the surgical management of CP has a biomechanical basis, surgeons find it easier to communicate with therapists who share this approach (80).
Biomechanical Approach.
The biomechanical approaches (80) aim to maintain range of motion and muscle length. Techniques used include manual passive ranging of joints, stretching of muscles, and splinting and casting. Serial casting can be combined with Botox injections, for dynamic contractures (84). The introduction of a suitable ankle foot orthosis is often a critical step in providing an improved base of support and achieving progress in a child progressing to standing and walking (38). Muscle weakness is a significant functional and biomechanical problem in children with CP. In children with hemiplegia, muscle weakness is also often compounded by nonuse of the affected limb (39). Progressive resistance strength training may be used to increase muscle strength and endurance (85).
Neurodevelopmental Therapy: The Bobath Approach.
Neurodevelopmental therapy (NDT) (81) is a widely used approach in children with CP. It aims to inhibit abnormal, primitive postural patterns and facilitate developmentally more mature movement patterns with improved function. NDT therapists use specific physical handling, including facilitation, to provide sensorimotor input and to guide motor output. Inhibitive casting is sometimes used with NDT to help achieve a reduction in tone and increased range of motion.
Cognitive Approach.
Cognitive or learning approaches (82) focus on learning control of movement for function rather than emphasizing quality of movement. Motor learning or training programs use task analysis to breakdown functional tasks into basic motor components or patterns. These components are then practiced and learned as a motor skill for functional use. Conductive Education (CE) or the Peto method is a cognitive approach that utilizes rhythm, music, and counting to initiate and moderate movements. CE is used extensively by therapists to develop skills necessary for the performance of daily activities such as dressing and self-feeding.
Constraint-Induced Movement Therapy.
CIMT (83) is used for learned nonuse of the affected upper limb, which is common in hemiplegia (39). It involves the forced use of the most affected upper limb by restricting use of the less involved hand in a cast, splint, or glove for periods of activity during the day.
The approach used by occupational therapists and physical therapists to manage children with CP is influenced by many factors. Intervention requires sensitivity to the child’s age, cognitive, sensory, and perceptual factors as well as type and severity of CP. The child’s environment, including family and culture, is important. Interventions will be less effective if they are not carried over into the child’s management at home. The treatment setting, whether based in a hospital, at school or home, and the focus of these services are also factors. The setting and the frequency of treatments are often influenced by the financial resources available.
Despite widespread use, evidence on the efficacy of physiotherapy to improve function in children with CP is equivocal. In randomized, controlled trials, treatment effects have generally been small.
MOVEMENT DISORDER MANAGEMENT AND THE SPASTICITY COMPASS
The “spasticity compass” can be used to classify and compare interventions for spasticity management, as focal or generalized in their effect and as temporary or permanent (84). Each intervention can then be located in the appropriate quadrant. For example, oral medications are general (all nerves or all muscles in all body areas) but temporary in effect (86). SDR is a neurosurgical procedure in which 30% to 50% of the dorsal rootlets between L1 and S1 are transected for the permanent relief of spasticity in a highly selected group of children with
spastic diplegia. The principal effects are on the lower limbs although there may be minor effects on the upper limbs. The position on the grid is therefore permanent and half way between general and focal (87, 88) (Fig. 14-10).
spastic diplegia. The principal effects are on the lower limbs although there may be minor effects on the upper limbs. The position on the grid is therefore permanent and half way between general and focal (87, 88) (Fig. 14-10).
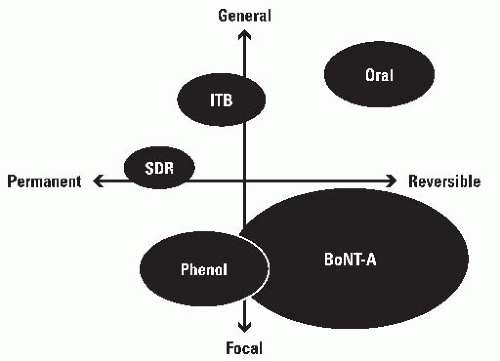 FIGURE 14-10. The spasticity compass as a guide to movement disorder management in CP. Interventions are classified on the north-south axis according to whether they are “general” or “focal” in their action. They are also classified on the east-west axis as to whether they are “permanent” or “reversible.” (From Gallagher C, Sheedy M, Graham HK. Integrated management with botulinum neurotoxin A. In: Panteliadis CP, ed. Cerebral palsy. A multidisciplinary approach. Munchen, Germany: Dustri-Verlag; 2011:213-236, with permission.) |
Oral medications used for the management of spasticity in children with CP include diazepam, baclofen, dantrolene sodium, and tizanidine. Artane and L-dopa are used in dystonia. All are limited in usefulness by a combination of limited benefits and side effects. They have been extensively reviewed in several recent publications (84, 89).
The limited lipid solubility of baclofen when administered orally can be overcome by intrathecal administration using a programmable, battery-operated implantable pump connected to a catheter and delivery system to the intrathecal space (86). This is an invasive procedure with associated morbidity and mortality (38, 39, 88). However, it is the most effective current method available for the management of severe spasticity, dystonia, and mixed movement disorders in CP and a number of other conditions including spasticity of spinal origin and acquired brain injury. The role of ITB has been reviewed extensively in several recent publications (86, 88).
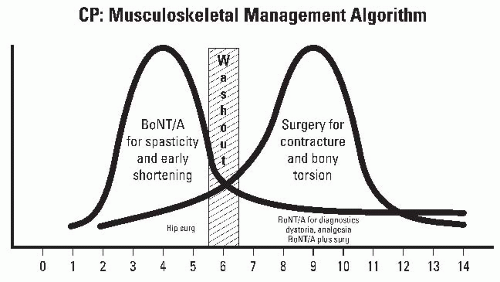 FIGURE 14-11. The CP musculoskeletal management algorithm. Neurolytic blocks, botulinum toxin A, and phenol are used in younger children with spasticity. Under the age of 6 years, the only surgery required is preventive hip surgery. Surgery for contracture and bony torsion is most often required between the ages of 6 and 12 years by which stage the role of Botulinum toxin is very limited. (From Gallagher C, Sheedy M, Graham HK. Integrated management with botulinum neurotoxin A. In: Panteliadis CP, ed. Cerebral palsy. A multidisciplinary approach. Munchen, Germany: Dustri-Verlag; 2011:213-236, with permission.) |
Chemodenervation is useful in the management of focal spasticity and dystonia. Phenol neurolysis was much more widely utilized before the introduction of Botulinum neurotoxin A (BoNT-A). The principal limitation on its use is pain at the site of injection and post injection dysesthesia. Phenol is not selective and has the same effect on sensory nerve fibers as motor fibers. The principal indications are neurolysis of the musculocutaneous nerve for elbow flexor spasticity and the obturator nerve for adductor spasticity. These nerves have limited sensory distribution (84, 90).
BOTULINUM NEUROTOXIN A IN CEREBRAL PALSY
Injection of skeletal muscle with BoNT-A results in a dosedependent, reversible chemodenervation, by blocking presynaptic release of acetylcholine at the neuromuscular junctions (84). Because of the toxin’s rapid and high-affinity binding to receptors at the neuromuscular junctions of the target muscle, little systemic spread occurs. Neurotransmission is restored first by sprouting of new nerve endings, followed by the original nerve endings regaining their ability to release acetylcholine (91, 92). BoNT-A may be useful in children with CP to manage dynamic gait problems and to delay the need for orthopaedic surgery until the child is older (Fig. 14-11).
Spastic Equinus.
The most common and most important indication for BoNT-A therapy in children with CP is the injection of the gastrocsoleus for spastic equinus (93, 94). Before widespread use of BoNT-A for spastic equinus, the majority of children with CP who walked on their toes had a lengthening of their Achilles’ tendons by age 4 to 6 years (95). This resulted in crouch gait that was much more disabling than the original equinus gait.
Now children with spastic equinus usually commence BoNT-A therapy aged between 1 and 3 years, in conjunction with physical therapy and the use of appropriate AFOs. They receive injections every 6 to 12 months for several years until gross motor function plateaus, at 4 to 6 years of age. Residual contractures and bony torsion can then be dealt with as SEMLS (96). A program of care utilizing BoNT-A should be viewed as complementary to surgical reconstruction and not as an alternative (96, 97) (Fig. 14-11). Information about dosing, dilution, muscle targeting, and safety has been published elsewhere (98, 99, 100, 101 and 102).
Now children with spastic equinus usually commence BoNT-A therapy aged between 1 and 3 years, in conjunction with physical therapy and the use of appropriate AFOs. They receive injections every 6 to 12 months for several years until gross motor function plateaus, at 4 to 6 years of age. Residual contractures and bony torsion can then be dealt with as SEMLS (96). A program of care utilizing BoNT-A should be viewed as complementary to surgical reconstruction and not as an alternative (96, 97) (Fig. 14-11). Information about dosing, dilution, muscle targeting, and safety has been published elsewhere (98, 99, 100, 101 and 102).
Injection of BoNT-A for spastic equinus increases the dynamic length of the gastrocsoleus with improvements in ankle dorsiflexion during gait, as determined by the Physician Rating Scale (93, 103). Improvements have been reported in studies using IGA, including kinematics, kinetics, and electromyography (94, 104). This may lead to small but important gains in gross motor function (105, 106 and 107). The evidence base supporting the use of BoNT-A in CP is quite good as confirmed in several randomized controlled trials and systematic reviews (108, 109). However, the treatment effect is small and short lived. The drug is expensive limiting access and is not approved by the FDA in the management of children with CP in the United States. All current use in the United States is therefore “off label.”
Spastic Equinovarus and Equinovalgus.
Spastic equinovarus is the result of spasticity in the gastrocsoleus, tibialis posterior and/or tibialis anterior (110). In spastic equinovarus, the most effective strategy is to inject the gastrocsoleus and the tibialis posterior (94). Equinovalgus is not usually the result of muscle imbalance but altered biomechanics. It is best managed by injection of the gastrocsoleus and provision of an appropriate AFO (109).
Injection of the Hamstrings and the Adductor Muscles in Cerebral Palsy.
Spasticity in the ham string and adductor muscles is prevalent in the severely involved child and may result in scissoring postures and spastic hip displacement (38). Injection of the adductor and hamstring muscles with BoNT-A every 6 months combined with an abduction brace had no appreciable effect on the prevention of hip displacement, in a large RCT (111). The majority of the children required surgical stabilization of their hips either during the study or soon after the study concluded (111).
Multilevel Injections of Botulinum Neurotoxin A in Cerebral Palsy.
Techniques have been developed for injecting the iliopsoas as part of a multilevel injection protocol for children with spastic diplegia. Multiple target muscles are injected under mask anesthesia and followed by supplemental casting, orthoses, and intensive rehabilitation. Temporary improvements in gait and function have been reported (112, 113). Phenol neurolysis for adductor spasticity can be combined with BoNT-A chemodenervation of the hamstring and calf muscles (90). The principal indication is the younger child with spastic hip displacement and walking difficulties (39, 84).
Botulinum Neurotoxin A in the Upper Limb in Cerebral Palsy.
In typical hemiplegic posturing the most common target muscles are biceps, brachialis, pronator teres, flexor carpi ulnaris, flexor carpi radialis, and adductor pollicis. The long finger flexors should usually be avoided to prevent weakening grip strength, except when the aim is improved palmar hygiene (92, 114, 115). Precise targeting with electrical stimulation, electromyography, or ultrasound is mandatory (101, 102). Palpation is inaccurate. Upper limb dose guides have been published elsewhere (92, 98, 99, 115). Botox injections in the upper limb results in a reduction in muscle tone but robust evidence for improvements in function is limited, in studies that employed valid and reliable functional outcome measures (116, 117, 118, 119, 120 and 121).
ADVERSE EVENTS AND BOTULINUM NEUROTOXIN THERAPY IN CEREBRAL PALSY
BoNT-A is generally safe in children with CP (93, 94, 98, 99). Most adverse events are localized, minor, and self-limiting. Systemic side effects including temporary incontinence and dysphagia have been reported (100). Dysphagia, aspiration, and chest infection are the most serious complications after injection of BoNT-A and if unrecognized or inadequately treated could lead to death from asphyxia (100, 122).
BOTULINUM NEUROTOXIN A AS AN ANALGESIC AGENT IN CEREBRAL PALSY
BoNT-A can be used to treat muscle spasm following operative procedures, such as adductor-release surgery (123). Injection of BoNT-A can be useful for short-term relief of pain associated with hip displacement (124). Target muscles include the hip adductors, medial hamstrings, and hip flexors. Pain relief is associated with a decrease in spastic adduction and scissoring postures (123, 124). It is not clear if short-term pain relief can be sustained by repeat injections and the need for salvage surgery avoided. Some children with neglected hip displacement have limited life expectancy and may not survive salvage surgery. BoNT-A may provide useful palliation in such circumstances (124). Better still is to prevent painful hip displacement.
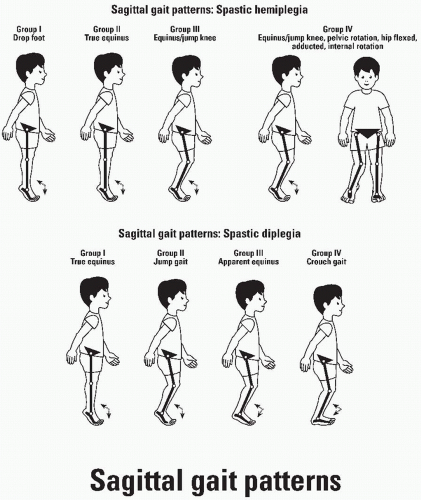
SAGITTAL GAIT PATTERNS: SPASTIC HEMIPLEGIA—WINTERS, GAGE, AND HICKS
In 1987, a four-group classification of sagittal gait patterns in spastic hemiplegia was developed by Winters et al. (125). Their four-group classification has been extensively used by
clinicians as a template for clinical management including prescription of orthoses, spasticity management by injection of BoNT-A, and musculoskeletal surgery (Fig. 14-12).
clinicians as a template for clinical management including prescription of orthoses, spasticity management by injection of BoNT-A, and musculoskeletal surgery (Fig. 14-12).
 |
Type I Hemiplegia.
In type I hemiplegia, there is a drop foot in the swing phase of gait due to loss of selective motor control in tibialis anterior. There is no contracture of the gastrocsoleus and second rocker is relatively normal. Neither spasticity management nor musculoskeletal surgery is necessary. Gait and function can be improved by the use of an AFO, usually a leaf spring AFO or a hinged AFO.
Type II Hemiplegia.
In type II hemiplegia, there is spasticity in the gastrocsoleus that gradually becomes fixed resulting in a contracture and equinus gait. First and second rockers at the ankle are disrupted and there may be proximal deviations related to the excessive plantar flexion at the ankle, but these are not primary gait deviations. Management of type II hemiplegia
requires correction of fixed contracture in the gastrocsoleus (to correct second rocker) and provision of an AFO (to provide heel strike and first rocker in stance as well as swing phase clearance and appropriate prepositioning of the foot during preswing).
requires correction of fixed contracture in the gastrocsoleus (to correct second rocker) and provision of an AFO (to provide heel strike and first rocker in stance as well as swing phase clearance and appropriate prepositioning of the foot during preswing).
Type III Hemiplegia.
In type III hemiplegia, there is a contracture of the gastrocsoleus at the ankle and knee involvement with co-contraction of the hamstrings and rectus femoris. Children in this transitional group may benefit from lengthening of the medial hamstrings and rectus femoris transfer (see Figs. 14-29, 14-30, 14-31, 14-32 and 14-33), in addition to gastrocsoleus lengthening.
Type IV Hemiplegia.
In type IV hemiplegia, pathology is present at all three joints of the lower extremity. In the sagittal plane, in addition to ankle equinus and knee stiffness, there is incomplete hip extension. In the coronal plane at the hip, there is excessive adduction and in the transverse plane, excessive internal rotation. Hip dysplasia is common and often presents late. In addition to the treatments outlined above for type III, correction of type IV hemiplegic gait includes lengthening of both the adductor longus and the psoas over the brim (POTB) of the pelvis as well as a proximal femoral derotation osteotomy.
SAGITTAL GAIT PATTERNS—SPASTIC DIPLEGIA
Knee patterns in spastic diplegia have been classified as recurvatum knee, jump knee, stiff knee, and crouch (128). The knee classification has been extended to the sagittal plane as true equinus, jump gait, apparent equinus, and crouch gait (97) (Fig. 14-12).
True Equinus.
True equinus is characterized by walking on tip toe with extended hips and knees, as is commonly seen in younger children with spastic diplegia when they first learn to walk. The plantarflexion-knee extension couple is overactive and the ground reaction force (GRF) is in front of the knee throughout stance phase. True equinus can be managed in the younger child by injections of BoNT-A to the gastrocsoleus and the provision of hinged AFOs. By the time children develop fixed contractures and require surgery, true equinus is rare. When it persists, there are usually occult contractures of the hamstrings and iliopsoas. Single-level surgery (gastrocsoleus lengthening) is almost never the correct strategy, no matter how tempting it may appear on observational gait analysis.
Jump Gait.
Jump gait is characterized by equinus at the ankle associated with incomplete extension at the knee and hip. In the original description by Sutherland and Davids, the jump knee pattern is characterized by excessive flexion at initial contact with rapid extension in later stance to near-normal range (128). In the pattern described by Rodda and Graham, jump gait encompasses this pattern as well as patterns in which knee extension is more severely compromised and in which there is incomplete extension at the hip (97, 127). This is the most common pattern in the preadolescent with spastic diplegia. Many children benefit from SEMLS.
Apparent Equinus.
Many children with spastic diplegia who walk on their toes, never achieving heel contact, have an ankle range of motion within the normal range. Such children are at risk of inappropriate management with injections of BoNT-A to the gastrocsoleus or even worse, lengthening of the gastrocsoleus. The important contractures are proximal at the level of the knee and hip. The recognition of “apparent equinus” in contradistinction to “true equinus” is very important to avoid inappropriate lengthening and weakening of the gastrocsoleus with further deterioration in gait and functioning. IGA is very helpful in differentiating “apparent equinus” from “true” equinus. Apparent equinus pattern is often transitional. With further growth and progression of lever arm deformities, the majority of children will eventually develop “crouch gait.”
Crouch Gait.
Crouch gait is characterized by excessive knee flexion in stance, incomplete extension at the hip, and excessive ankle dorsiflexion. Knee stiffness in swing is common. The soleus is excessively long and usually weak. This is a very common gait pattern in adolescence and is often the result of natural history, accelerated by lengthening of the gastrocsoleus, especially percutaneous lengthening of the Achilles tendons. In recent reviews of crouch gait, the majority of children had lengthening of the Achilles tendons in childhood (59). A key feature of crouch gait is that the majority of MTUs are excessively long. This is by definition true for all of the one joint muscles such as soleus, quadriceps, and gluteus maximus and often for the two joint hamstrings. The only consistent contractures are of the iliopsoas. In crouch gait, the hamstrings are short only in patients with a posterior pelvic tilt. When the pelvis is in the neural range, the hamstrings are of normal length and when the pelvis is anteriorly tilted, the hamstrings are excessively long. Without the use of IGA and the plotting of muscle lengths, it is very difficult to appreciate these findings. Consequently, the majority of children with crouch gait are managed by excessive hamstring lengthening to improve knee extension when in fact the hamstrings are of normal length or excessively long. Such surgery results in increased anterior pelvic tilt that in the long term may bring its own set of problems with low back pain and increased risks of spondylolisthesis and spondylolysis (59).
BIOMECHANICAL STUDIES RELEVANT TO ORTHOPAEDIC SURGERY IN SPASTIC DIPLEGIA
In a study of muscle excursion and cross-sectional area, it was demonstrated that the gastrocsoleus and ankle dorsiflexors have such different physical characteristics that they cannot
be considered to be “in balance,” in either normal subjects or in children with CP (129). The plantarflexors are six times as strong as the dorsiflexors. The plantarflexors of the ankle must be balanced against the GRF not the dorsiflexors. Lifting the foot and ankle during swing phase (dorsiflexor function) requires a very small muscle moment. Push-off in terminal stance (plantarflexor function) requires a large muscle moment. The concept of muscle balance should be redefined as a requirement for balance between the three anatomical levels, hip, knee and ankle, in the sagittal plane, not at a single level (129).
be considered to be “in balance,” in either normal subjects or in children with CP (129). The plantarflexors are six times as strong as the dorsiflexors. The plantarflexors of the ankle must be balanced against the GRF not the dorsiflexors. Lifting the foot and ankle during swing phase (dorsiflexor function) requires a very small muscle moment. Push-off in terminal stance (plantarflexor function) requires a large muscle moment. The concept of muscle balance should be redefined as a requirement for balance between the three anatomical levels, hip, knee and ankle, in the sagittal plane, not at a single level (129).
Lower limb muscles have different sensitivities to surgical lengthening, related to their gross anatomy and morphology. The soleus is exquisitely sensitive to lengthening, but the iliopsoas and semitendinosus are relatively resistant. A 1-cm lengthening of the soleus reduces its moment-generating ability by 30% and a 2-cm lengthening reduces its moment by 85% (130). A small error in terms of overlengthening the soleus may be disastrous. A 4-cm lengthening of the psoas is required to reduce its moment by 50% (130). The surgical implications are to lengthen the gastrocnemius only, when there is no contracture of the soleus. When the soleus requires lengthening, a precise and stable technique should be used, with careful control of the position postoperatively in a cast. By contrast, it is difficult to overlengthen the psoas. Intramuscular lengthening at the pelvic brim without immobilization postoperatively is safe and effective.
MUSCULOSKELETAL MANAGEMENT IN CEREBRAL PALSY BY GMFCS LEVEL: INTRODUCTION
The GMFCS gives an accurate summary of a child’s current gross motor function and long-term prognosis (13, 15). It is difficult to frame a logical discussion of musculoskeletal management outside of the context of the GMFCS. Most disagreements in the literature are apparent rather than real because those taking opposing views are often considering a child in a different GMFCS level. The terms mild, moderate, and severe diplegia along with mild, moderate, and severe quadriplegia are not meaningful or useful. Age-appropriate GMFCS descriptors are valid, reliable, stable, and clinically meaningful (47, 49, 50). Recommendations for both gait correction surgery and surgery for hip displacement are much more easily understood when the child’s GMFCS level is known. In the following sections, musculoskeletal management will be discussed by GMFCS level, in conjunction with both topographical classification and sagittal gait patterns. Gait correction surgery is an option for many children with CP at GMFCS levels I to III but with differences at each level. SEMLS will be discussed primarily in the GMFCS level II section. Preventive hip surgery will be discussed in the GMFCS level III section, reconstructive hip surgery in the GMFCS level IV section, and salvage surgery in the GMFCS level V section. Obviously, reconstructive surgery may be appropriate at GMFCS levels III to V and salvage surgery is sometimes needed at both GMFCS IV and V but is best avoided.
GMFCS I
1. Between 6th and 12th birthday
Children walk at home, school, outdoors, and in the community. They can climb stairs without the use of a railing. Children perform gross motor skills such as running and jumping, but speed, balance, and coordination are limited (13).
2. Between 12th and 18th birthday
Youth walk at home, school, outdoors, and in the community. They are able to climb curbs and stairs without physical assistance or a railing. They perform gross motor skills such as running and jumping, but speed, balance, and coordination are limited (15).
3. Risk of hip displacement: Developmental dislocation of the hip occurs at the same rate as in the normally developing population. Spastic hip displacement is not seen (49, 50).
4. Mean femoral neck anteversion (FNA): 30 degrees
5. Mean neck shaft angle (NSA): 136 degrees
Children at GMFCS level I participate in physical recreational activities, have a normal life expectancy, few medical comorbidities apart from epilepsy, and good levels of intellectual functioning.
Movement Disorder.
Topographical Distribution.
Children at GMFCS level I have either spastic hemiplegia or mild spastic diplegia. In terms of sagittal gait patterns, those with hemiplegia are usually type I or II. Those with diplegia usually have true equinus or mild jump gait.
Musculoskeletal Impairments.
Hip disease and scoliosis are rare and occur with a similar prevalence as would be expected in typically developing children. If hip displacement is detected, it is usually a developmental dysplasia. If a spinal curvature develops, it will be an adolescent idiopathic-type curve.
Musculoskeletal Management GMFCS Level I: Spastic Hemiplegia
Lower Limb Surgery.
Children with type I hemiplegia have a drop foot in the swing phase of gait. They may benefit from a leaf spring or a hinged AFO. Orthopaedic surgery is not required (125).
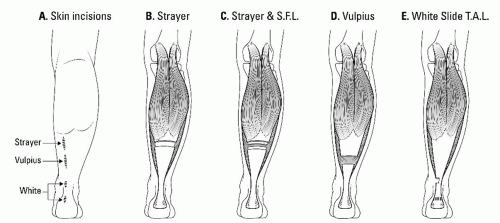 FIGURE 14-13. Surgery for equinus deformity in cerebral palsy. A: The location of the skin incisions is usually posteromedial and with accurate identification of the level can be kept small, typically 2 to 3 cm for the Strayer and Vulpius procedures. The White slide TAL may be performed percutaneously, but our preference is for two small posteromedial incisions each 1.5 cm long. We prefer to see the tendon and avoid accidental complete tenotomy. B: The Strayer procedure is a distal gastrocnemius recession and lengthens only the gastrocnemius portion of the gastrocsoleus. It is the most useful procedure in children with diplegia and “gastrocnemius equinus.” C: The Strayer procedure can be combined with soleal fascial lengthening (SFL). This results in a lengthening of both gastrocnemius and soleus but by different amounts, a 2:1 ratio, that is, twice as much lengthening of the gastrocnemius as for the soleus. This is the most useful procedure in children with spastic diplegia who have a moderate contracture of the gastrocnemius and a less severe contracture of the soleus. D: The Vulpius procedure is a Zone 2 recession of the gastrocsoleus. The shape of the cut can be the familiar inverted V. However, a simple transverse cut requires a smaller skin incision and is just as effective. This is a useful procedure in children with hemiplegia who have a moderate degree of fixed contracture affecting both the gastrocnemius and the soleus. E: Slide lengthening of the Achilles tendon may be performed by double hemisection as described by White. This is the most useful procedure in children with hemiplegia who have a severe contracture affecting the gastrocnemius and the soleus. For further discussion please see reference 133. |
Children with type II hemiplegia develop equinus contractures and may benefit from lengthening of the gastrocsoleus. Surgery can usually be deferred until age 4 to 6 years by the use of an AFO combined with injections of BoNT-A and physical therapy. The choice of lengthening procedure is based on a careful Silfverskiold test to determine the amount of contracture in the gastrocnemius and soleus, respectively (133). There are four main options as illustrated in Figure 14-13 (see Figs. 14-14 and 14-15).
Upper Limb Surgery in Spastic Hemiplegia.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


