Chapter 27 Cardiovascular Disease
Dyslipidemia
Causes
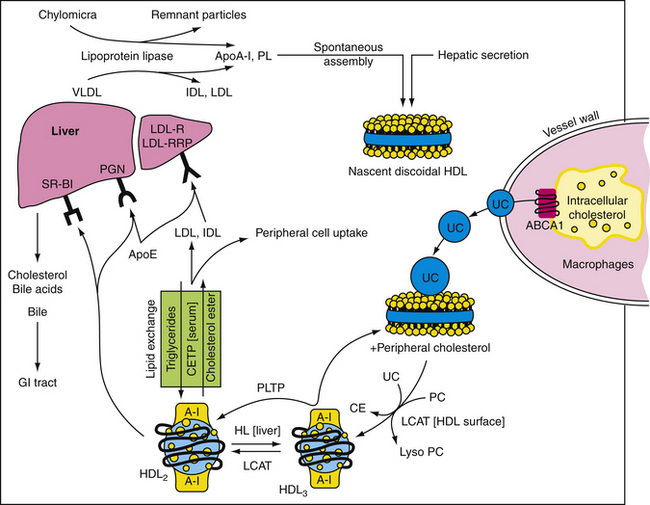
High-density lipoprotein (HDL) particles are antiatherogenic. With few exceptions, epidemiologic investigation has shown that high HDL levels reduce risk for the development of CAD (Toth, 2001, 2009). Consistent with this finding, patients with familial hypo-α-lipoproteinemia (low HDL) have increased risk for premature CAD, whereas patients with familial hyper-α-lipoproteinemia (high HDL) are relatively resistant to CAD (Toth, 2003, 2004). HDL extracts excess intracellular cholesterol from macrophages and delivers it back to the liver for elimination as bile salts through the gastrointestinal tract in a process referred to as “reverse cholesterol transport” (Fig. 27-1). HDL has also been shown to reduce endothelial CAM expression, augment endothelial nitric oxide and prostacyclin production, reduce oxidized LDL, decrease platelet aggregability, and inhibit endothelial cell apoptosis (programmed cell death). An HDL greater than 60 mg/dL is a “negative” risk factor. In general, the more elevated the level of serum HDL, the lower is the risk for CAD. Therapeutic interventions should not be targeted at reducing serum HDL. Although emerging evidence suggests that some patients may harbor HDL species that appear to be proinflammatory and pro-oxidative because of alterations in their protein and enzyme cargo (Ansell, 2006), treatment with some types of lipid-modifying agents appears to restore normal functionality to these HDL particles (Heinecke, 2009).
Risk Factors
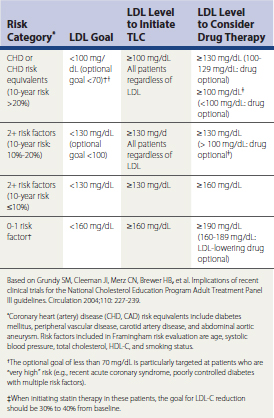
The National Cholesterol Education Program Adult Treatment Panel III (NCEP ATP-III, 2001) has defined risk-stratified, evidence-based target levels for atherogenic serum lipoproteins (Table 27-1). CAD risk is stratified by evaluating a patient’s cardiovascular risk factor burden (number of risk factors) and, if two or more risk factors are present, calculation of the Framingham risk score. In the setting of primary prevention, low risk is defined as 0-1 risk factor. Moderate and moderately high risk are defined as 2 or more risk factors with a 10-year Framingham risk of 5% to 10% and 10% to 20%, respectively. Patients who are in the high risk category either have CAD (previous history of MI, stable/unstable angina, revascularization with CABG or percutaneous coronary angioplasty) or a CAD risk equivalent, defined as diabetes mellitus, abdominal aortic aneurysm, peripheral vascular disease, significant carotid artery disease (transient ischemic attack or stroke from carotid origin, >50% obstructive atheromatous plaque in carotid artery), or a 10-year Framingham risk that exceeds 20%. The American Heart Association (AHA) also defines chronic kidney disease (glomerular filtration rate [GFR] <60 mL/min/1.73 m2) as a CAD risk equivalent. It is important to calculate the Framingham risk score so as to differentiate moderate, moderately high, and high risk among patients with multiple risk factors and no history of CAD or a CAD risk equivalent. An electronic Framingham risk calculator can be downloaded at www.nhlbi.nih.gov/guidelines/cholesterol. Box 27-1 summarizes established CAD risk factors.
Table 27-1 Low-Density Lipoprotein (LDL) Cholesterol Goals and Thresholds for Initiating Therapeutic Lifestyle Change (TLC) and Pharmacologic Intervention
Targets
In ATP-III the NCEP implemented these conceptual changes: (1) an optimal LDL-C is defined as less than 100 mg/dL for all patients independent of race or gender; (2) an HDL less than 40 mg/dL is now defined as a categorical risk factor for CAD; and (3) it defined target levels for non-HDL-C. Non-HDL-C is defined as total cholesterol minus HDL-C and is an estimate of atherogenic lipoproteins in serum (VLDL + LDL). The risk-stratified target for non-HDL-C is the LDL-C target plus 30 (see Table 27-1). LDL-C reduction is the primary goal of therapy in patients with dyslipidemia. However, in patients with fasting triglyceride (TG) levels greater than 200 mg/dL, non-HDL reduction is the secondary priority of therapy. There is currently no specified target for HDL-C elevation. However, in patients with low HDL-C, it is important to try to raise HDL-C as much as possible. According to a recent AHA Consensus Statement, an HDL-C less than 50 mg/dL in women is now considered low (Mosca et al., 2004). The American Diabetes Association (ADA) advocates HDL-C goals of 40 mg/dL or higher for men and 50 mg/dL or higher for women with diabetes mellitus.
Nonpharmacologic Interventions
Therapeutic lifestyle change (TLC) is first-line therapy for patients at risk for cardiovascular events. Patients who smoke should stop. The amount of daily consumed cholesterol should be less than 200 mg. Table 27-2 summarizes the distribution of calories from nutrients. Reduced saturated fat and increased consumption of mono- and polyunsaturated fats promote serum LDL-C reduction. The ingestion of viscous fiber and plant stanols decrease cholesterol absorption. Ideally, patients should exercise for 20 to 30 minutes five times per week. Regular exercise promotes weight loss and relieves visceral adiposity and insulin resistance.
Table 27-2 Dietary Recommendations for Therapeutic Lifestyle Change
| Dietary Component | Recommended Allowance |
|---|---|
| Polyunsaturated fat | Up to 10% of total calories |
| Monounsaturated fat | Up to 20% of total calories |
| Total fat | 25%-35% of total calories |
| Carbohydrate | 50%-60% of total calories |
| Dietary fiber | 20-30 g/day |
| Protein | About 15% of total calories |
| Dietary cholesterol | <200 mg/day |
Pharmacologic Interventions
Statins
The statins display significant differences in their pharmacokinetic profiles. Because of their relatively short half-life (1-4 hours), lovastatin, fluvastatin, pravastatin, and simvastatin should be taken after the evening meal in order to intercept the peak activity of HMG–CoA reductase, which occurs around midnight. Rosuvastatin and atorvastatin can be taken at any time during the day or night because of their long half-life (~19 and 14 hours, respectively). The coadministration of drugs or compounds that inhibit cytochrome P450 3A4 (macrolide antibiotics [erythromycin, clarithromycin], azole-type antifungals [ketoconazole, itraconazole], cyclosporine, HIV protease inhibitors, nefazodone, >1 qt grapefruit juice daily) with atorvastatin, simvastatin, and lovastatin is contraindicated because these statins depend on this P450 isozyme for oxidative modification and elimination (Neuvonen et al., 1998). CYP3A4 inhibition is associated with increased risk for myopathy and hepatotoxicity. The dose of simvastatin should be 20 mg or less daily in patients being treated with amiodarone or verapamil.
Ezetimibe
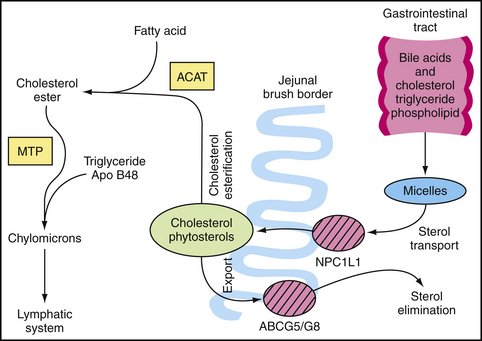
Dietary and biliary sources contribute significantly to serum levels of cholesterol (Fig. 27-2). Although plant sterols and stanols block gastrointestinal (GI) cholesterol absorption, ezetimibe (Zetia) is the first member of a class of lipid-lowering drugs known as cholesterol absorption inhibitors. Mechanistically, ezetimibe inhibits the Niemann-Pick C1 Like-1 protein, which mediates cholesterol and phytosterol transport along the brush border of the jejunal enterocyte (Altmann et al., 2004; Davis et al., 2004). After glucuronidation, ezetimibe undergoes enterohepatic recirculation with negligible systemic exposure. The half-life of ezetimibe is approximately 22 hours. When dosed at 10 mg once daily, ezetimibe reduces serum LDL-C on average by 20%, but up to 24% of patients experience a reduction of 25% or greater (Ballantyne et al., 2004; Davidson et al., 2002). Ezetimibe also decreases TGs by up to 8% and raises HDL-C by up to 4%. Ezetimibe does not decrease the absorption of bile acids, steroid hormones (ethinyl estradiol, progesterone), or such fat-soluble vitamins as vitamins A, D, E, or α- and β-carotenes.
Bile Acid–Binding Resins
The bile acid sequestration agents (BASAs) are orally administered anion-exchange resins that bind bile acids in the GI tract and prevent them from being reabsorbed into the enterohepatic circulation. These drugs reduce serum LDL-C by two mechanisms: (1) increased catabolism of cholesterol secondary to the upregulation of 7-α-hydroxylase, the rate-limiting enzyme for the conversion of cholesterol into bile acids; and (2) increased expression of LDL receptors on the hepatocyte surface, which augments the clearance of apoB100-containing lipoproteins from plasma. At maximum doses, the BASA can reduce serum LDL-C by 15% to 30% and increase HDL-C by 3% to 5%. It is recommended that these drugs be used in conjunction with a statin whenever possible because BASA therapy increases HMG-CoA reductase activity in the liver, which leads to increased hepatic biosynthesis of cholesterol, thereby offsetting the effects of the BASA over time. The BASAs are contraindicated in patients with serum TG levels greater than 400 mg/dL because these agents can exacerbate hypertriglyceridemia.
Fibrates
As with the statins, fibrates are associated with a low incidence of myopathy and mild elevations in serum transaminases. Fibrate therapy can increase the risk for cholelithiasis and elevate prothrombin time by displacing warfarin from albumin-binding sites. The periodic monitoring of serum transaminases (6-12 weeks after initiating therapy and twice annually thereafter) is recommended. The two most common fibrates are gemfibrozil (Lopid; 600 mg twice daily) and fenofibrate (Tricor; 54 or 160 mg daily). Bezafibrate is available in Europe and is dosed at 400 mg daily. The use of therapies combining a statin and fibrate is becoming more commonplace in clinical practice, especially as the incidence of complex dyslipidemias increases (Davidson and Toth, 2004). Gemfibrozil significantly reduces the glucuronidation of statins, which decreases their elimination (Backman et al., 2002; Prueksaritanont et al., 2002a, 2002b). This increases the risk for myopathy/rhabdomyolysis and hepatotoxicity. When used in combination with gemfibrozil, the doses for simvastatin and rosuvastatin should not exceed 10 mg daily. In general, when embarking on combination therapy, fenofibrate is a safer choice because it does not adversely impact the glucuronidation of the statins (Bergman et al., 2004). Fenofibric acid (Trilipix) is indicated for use in combination with statin therapy. Although there are no clinical trial data yet available to assess the effect of statin-fibrate combination therapy on cardiovascular morbidity and mortality, the efficacy of fenofibrate used in combination with simvastatin compared with simvastatin monotherapy is being tested in diabetic patients in the Action to Control Cardiovascular Risk in Diabetes Trial (http://www.nhlbi.nih.gov/health/prof/heart/other/accord/).
Fish Oils
Fish oil capsules enriched with omega-3 (eicosapentaenoic acid) and omega-6 (docosahexaenoic acid) fatty acids can reduce serum triglyceride and VLDL levels and raise HDL-C in a dose-dependent manner. The omega-3 fatty acids inhibit the enzyme diacylglycerol acyltransferase-2, thereby reducing intrahepatic TG biosynthesis. They also stimulate mitochondrial beta-oxidation of fatty acids, decrease VLDL production and biosynthesis, and stimulate TG hydrolysis by LPL (Toth et al., 2009). Dietary supplementation with the n-3 polyunsaturated fatty acids (PUFAs) eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) has also been shown to lower the risk of death, nonfatal coronary events, and stroke after MI (GISSI-Prevenzione Investigators, 1999). In several clinical trials, PUFAs have been shown to reduce TG levels by 20% to 30%, and up to 50% in patients with severe hypertriglyceridemia (TG >500 mg/dL) (O’Keefe and Harris, 2000).
Niacin
KEY TREATMENT
Potential for Harm
Hypertension
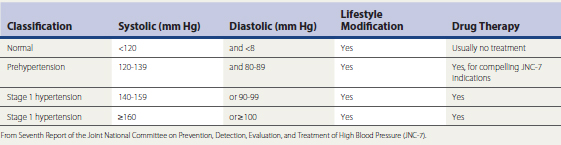
Hypertension (HTN) is highly prevalent and is a significant risk factor for CAD, left ventricular hypertrophy (LVH), CHF, PVD, stroke, sudden death, nephropathy, and diabetes mellitus. The incidence of HTN increases as a function of age; patients who are normotensive at age 55 have a 90% risk of developing HTN at some point in their lives. Risk for hypertension is regulated by genetic background (e.g., mutations in cell membrane cation transporters in the renal proximal tubular epithelium and vasculature, cell surface receptors, endocrine influences, calcium handling in smooth muscle cells) and environmental factors. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC-7) provides the current framework for defining and managing HTN in the U.S. population (Chobanian et al., 2003). Table 27-3 summarizes the JNC-7 classification of blood pressure (BP), soon scheduled for revision. The reduction of BP to guideline-specified targets in patients with complicated and uncomplicated HTN substantially reduces risk for acute cardiovascular events, progression of atherosclerosis, and end-organ injury.
Recommendations
A large number of antihypertensive medications are available (Table 27-4). Prevailing expert opinion now contends that, unless there are compelling indications for the use of a specific drug class because of background cardiovascular or renal disease, it makes little difference whether a calcium channel blocker (CCB), beta blocker, angiotensin converting enzyme inhibitor (ACEI), or angiotensin receptor blocker (ARB) is used to initiate therapy in the setting of uncomplicated HTN (Blood Pressure Lowering Treatment Trialists wwwwCollaboration, 2003; Julius et al., 2004; National Institute of Clinical Excellence Guideline 18, 2004; Pepine et al., 2003; Williams, 2005). Risk reduction is predominantly driven by the magnitude of BP reduction, rather than the specific mechanism by which it is achieved. Ideally, pharmacologic intervention will be coupled to lifestyle modification in order to achieve lasting and consistent reductions in BP reduction. If a patient’s BP is greater than 20/10 mm Hg above goal, initiating therapy with a combination preparation increases the likelihood of therapeutic success and patient compliance. The use of combination therapy is also associated with reduced cost. Many ACEI, ARBs, CCBs, centrally acting drugs (e.g., reserpine, methyldopa), and β-blockers are available in combination with fixed doses of hydrochlorothiazide (12-25 mg) and, less often, chlorthalidone. Box 27-2 summarizes combinations of drugs that are efficacious and those associated with undesirable side effect profiles. Box 27-3 summarizes recommendations for attaining BP targets in the more challenging patient.
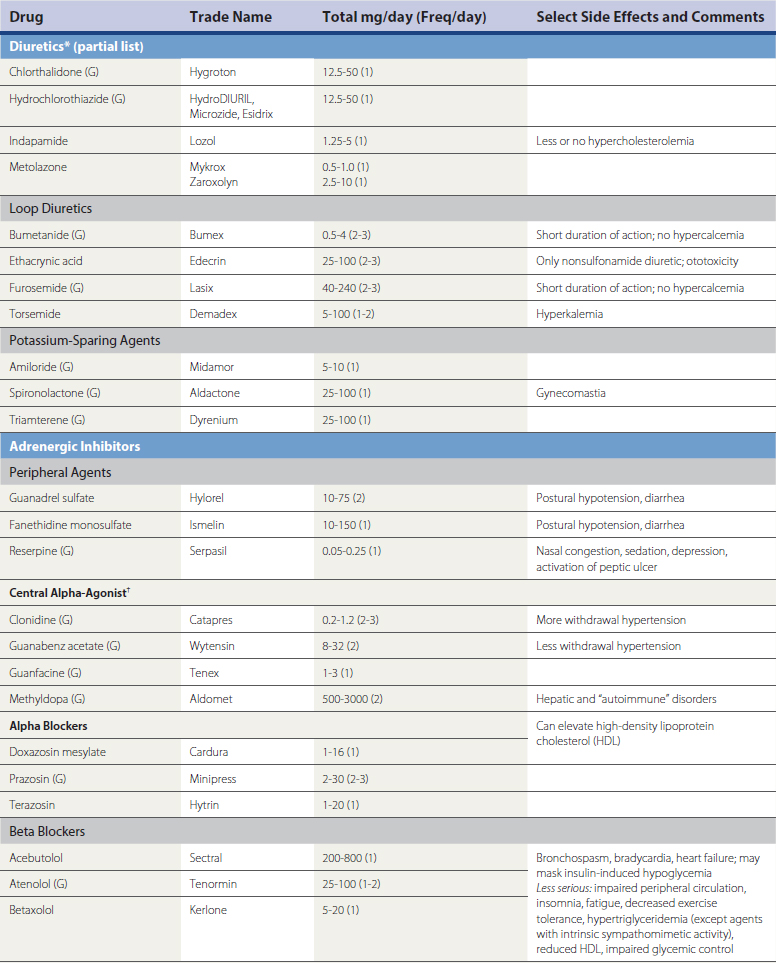
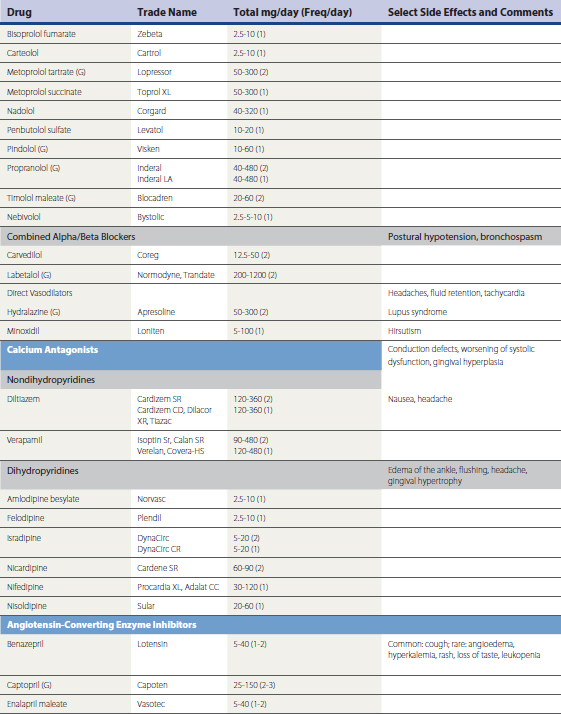
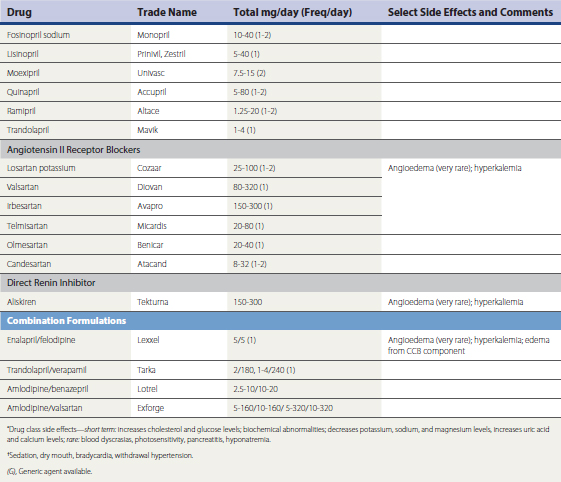
Box 27-3 Ten Tips for Attaining Goal Blood Pressure (BP)
Modified from Flack JM, Nasser SA. Hypertension Pocket Guide. New York, McMahon, 2005.
Patients with BP poorly responsive to even aggressive intervention should be evaluated further for etiologies such as renal arterial stenosis, adrenal and pituitary tumors, poor compliance, thyroid “storm,” and volume overload from acute or chronic renal disease. A broad range of clinical trials have demonstrated that the rate of progression of certain cardiovascular and renal disease states is slowed by the use of particular antihypertensive agents (Chobanian et al., 2003). These constitute “compelling indications” (Table 27-5). In patients requiring inhibition of the renin-angiotensin-aldosterone (RAAS) axis (CHF, CAD, post-MI, nephropathy, LVH), an elevation in serum creatinine of up to 35% is tolerable and is not an indication for discontinuing an ACEI or an ARB. In patients who develop hyperkalemia in response to an ACEI or ARB therapy, consideration should be given to reducing their dosage, adding a thiazide diuretic to promote potassium excretion, reducing potassium intake, or, when necessary, discontinuing the drugs and arranging referral to a nephrologist. Acute hyperkalemia can be managed with oral or rectal Kayexalate, a resin that binds and promotes the excretion of GI potassium. Men with HTN and benign prostatic hypertrophy or low serum HDL can be treated with an alpha-adrenergic blocker. In contrast to α-blockade, which can raise HDL levels by up to 20%, β-blockade is associated with reductions in serum HDL. Beta blockers and thiazide diuretics are both associated with the antagonism of glycemic control. Potassium balance should be monitored regularly in patients receiving combinations of ACEI/ARB with aldosterone antagonists. Blood pressure management should be individualized, with providers paying close attention to potential unwanted side effects that may dampen the benefit of BP reduction.
Table 27-5 JNC-7–Defined Compelling Indications for Use of Specific Antihypertensive Agents in Complicated Hypertension
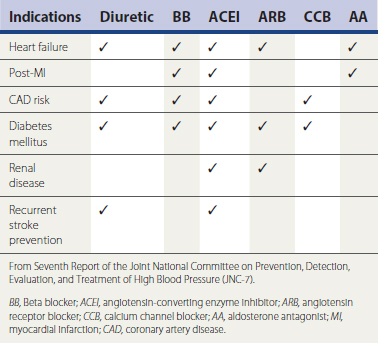
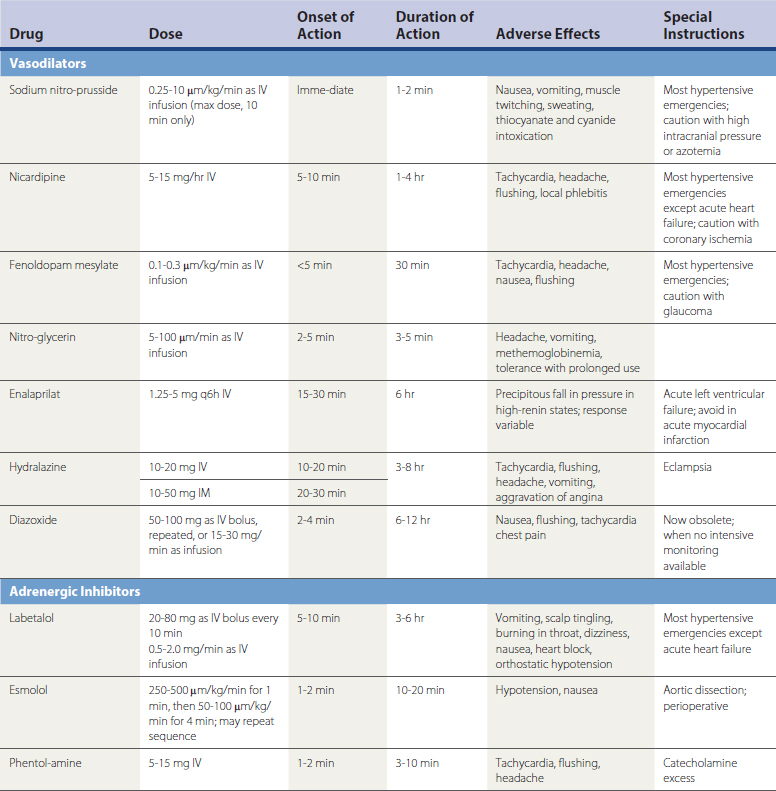
Hypertensive emergencies increase risk for acute MI, hemorrhagic stroke, encephalopathy, and other adverse events. Table 27-6 summarizes intravenous (IV) medications that can be used to reduce BP rapidly in the acute, emergent setting.
The results of two prospective trials may impact JNC-8 recommendations. In the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT) an atenolol/hydrochlorothiazide (A/H) regimen was compared to amlodipine/perindopril (A/P) in 19,257 subjects with HTN and three other cardiovascular risk factors. Despite nearly identical BP control, the patients randomized to A/P experienced a 23% lower incidence of fatal/nonfatal stroke, 16% fewer cardiovascular events and procedures, 11% lower all-cause mortality, and 30% less new-onset type 2 diabetes mellitus compared to the A/H regimen over a median follow-up of 5.5 years (Dahlof et al., 2005). In the Avoiding Cardiovascular Events through Combination Therapy in Patients Living with Systolic Hypertension trial, 11,506 hypertensive patients were randomized to treatment with either benazepril/amlodipine (B/A) or benazepril/hydrochlorothiazide (B/H). The primary and secondary composite end points comprised (1) nonfatal MI and stroke, hospitalization for angina, resuscitation after sudden cardiac arrest, coronary revascularization, and cardiovascular mortality; and (2) nonfatal MI and stroke and cardiovascular mortality, respectively (Jamerson et al., 2008). The primary and secondary end points were both reduced more with B/A compared to B/H by 19.6% and 21%, respectively. These trials strongly support the use of ACE inhibition therapy combined with the dihydropyridine amlodipine when managing hypertension. Among patients with established vascular disease or high-risk diabetes mellitus without heart failure, the ONTARGET trial demonstrated that the incidence of cardiovascular events was identical in groups treated with either telmisartan or ramipril (Yusuf et al., 2008). The incidence of angioedema in the telmisartan group was approximately one third of that observed in the ramipril group. Consequently, in patients such as those studied in ONTARGET, it is therapeutically legitimate to treat ACEI-intolerant patients with an ARB.
KEY TREATMENT
Potential Harm
Metabolic Syndrome
Causes and Incidence
The incidence of obesity is rising worldwide. With increased mechanization and changes associated with increased food availability and lower average daily caloric expenditure, people are experiencing continuous weight gain with aging. Being overweight and the development of obesity now constitute the second most important preventable cause of mortality. In the United States, 30% of the population is obese, and 70% is overweight. The incidence of obesity is rising among both genders as well as all racial and ethnic groups. An important consequence of obesity is the development of insulin resistance and the metabolic syndrome (Haffner et al., 1990; Haffner and Taegtmeyer, 2003). The metabolic syndrome (or “syndrome X”) is defined by a constellation of cardiovascular risk factors and is associated with heightened risk for cardiovascular morbidity and mortality. A variety of definitions of the metabolic syndrome have been developed, but the one with the greatest clinical utility is that defined by the NCEP ATP-III (Grundy et al., 2004a, 2004b; Eckel et al., 2005). Waist circumference, blood pressure, fasting blood sugar, and serum TG and HDL levels are used to make the diagnosis (Table 27-7). Once three of the five criteria are met, the diagnosis of metabolic syndrome can be made.
Table 27-7 NCEP ATP-III Criteria for Diagnosing Metabolic Syndrome
| Risk Factor | Defining Level |
|---|---|
| Abdominal obesity | |
| Triglycerides | ≥150 mg/dL |
| High -density lipoprotein cholesterol (HDL-C) | |
| Blood pressure | ≥130/ ≥85 mm Hg |
| Fasting glucose | ≥100 mg/dL |
∗Patients having any three of the above five risk factors meet criteria for the diagnosis of the metabolic syndrome.
From Third Report of the Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) (NCEP ATP-III).
Cardiovascular Risk Factors
As waist circumference increases, visceral adiposity increases. Visceral adipose tissue is metabolically highly active. An important conceptual shift has occurred in recent years with respect to how adipose tissue is viewed. It is no longer seen as a passive storage site for excess caloric ingestion. Instead, it is clear that visceral adipose tissue displays many features of an endocrine organ (Bradley et al., 2001; Toth, 2005a) (Fig. 27-3). Visceral adipose tissue produces a variety of inflammatory cytokines (tumor necrosis factor, transforming growth factor-β), interleukins (IL-1, IL-6), and effector molecules that regulate appetite (leptin) as well as insulin sensitivity and resistance (e.g., adiponectin, resistin). As the mass of visceral adipose tissue increases, adiponectin production decreases, which is associated with increased insulin resistance in adipose tissue, skeletal muscle, and the hepatic parenchyma (see Fig. 27-3). As adipose tissue becomes more insulin resistant, the capacity to regulate the catabolism of stored TGs becomes progressively more dysregulated and unresponsive to systemic tissue needs. Serum levels of free fatty acids (FFAs) rise. The portal circulation becomes flooded with FFAs, resulting in both increased TG deposition within the liver (nonalcoholic steatohepatitis [NASH] or fatty liver) (Banerji et al., 1995) and increased VLDL secretion resulting in hypertriglyceridemia. A fatty liver in the absence of excessive alcohol intake is an important marker for insulin resistance and is highly correlated with the magnitude and severity of adiposity (Fig. 27-4). Elevation in FFAs induces progressive deterioration in glycemic control by (1) interfering with normal phosphorylation of the insulin receptor, resulting in less expression of a glucose transporter (GLUT 4) necessary for the internalization and oxidation of serum glucose (Dresner et al., 1999), and (2) the induction of “lipotoxicity,” the process by which FFAs induce premature apoptosis and dropout of pancreatic β-islet cells. Patients experiencing concomitant worsening insulin resistance and progressive loss of insulin-producing capacity experience a continuum of glycemic disturbance, beginning with impaired fasting glucose, then impaired glucose tolerance, and ultimately diabetes mellitus. As serum levels of insulin rise, risk for CAD-related events increases precipitously (Pyorala et al., 1996) (Fig. 27-5). Patients with metabolic syndrome have a threefold to fivefold increased risk for developing diabetes compared to patients without metabolic syndrome.
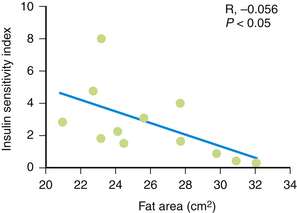
Figure 27-3 Insulin sensitivity and degree of adiposity.
(From Fujimoto WY, Abbate SL, Kahn SE, et al. The visceral adiposity syndrome in Japanese-American men. Obes Res 1994;2:364-371.)






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


