Chapter 33 Inherited cancer syndromes


Genetic mutations and disease development
Neoplastic diseases are characterized by the presence of genetic mutations within the neoplastic tissue (Fig. 3.33.1), which are often multiple in nature and which are associated with loss of the normal mechanisms controlling cellular growth and maturation (Ch. 22). In the majority of cases, cells within the affected individual’s non-neoplastic tissue do not usually contain demonstrable genetic mutations (i.e. they appear genetically normal).
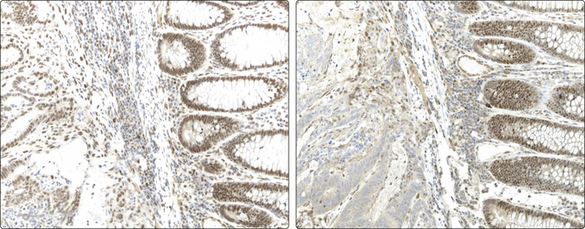
Fig. 3.33.1 Photomicrographs of a colorectal cancer from a patient with the hereditary non-polyposis colorectal cancer (HNPCC) syndrome in which immunohistochemistry shows expression of the DNA mismatch repair enzyme hMLH-1 (brown stain within tumour cell nuclei) but loss of expression of the DNA mismatch repair enzyme hMSH-2 (the negative tumour cell nuclei show only the blue counterstain). In both, the tumour is on the left side and normal large bowel mucosa (serving as an internal positive control for the staining method) is on the right side. This patient has an inherited mutation within the gene encoding hMSH-2.
Inherited predisposition to neoplastic disease
Inherited cancer syndromes are characterized by the presence of a mutation that is inherited from one or both parents and which leads to a significantly increased risk of development of a particular neoplasm or group of neoplasms. This is reflected in an increased incidence of neoplasia within affected families, usually with an identifiable Mendelian pattern of inheritance in a dominant fashion (Table 3.33.1). Inherited mutations associated with familial cancer syndromes are usually present within genes important in the regulation of cellular growth and/or maturation and are commonly termed tumour suppressor genes (Ch. 22). Neoplasms develop when mutation or loss of the second normal copy of the gene occurs such that the tumour tissue then contains no normal copies of the gene.
Table 3.33.1 INHERITED NEOPLASIA SYNDROMES
| Syndrome | Gene | Clinical manifestation |
|---|---|---|
| Familial adenomatous polyposis | APC (adenomatous polyposis coli) | Multiple adenomas in colon and small intestine; colorectal cancer at very young age |
| Hereditary non-polyposis colorectal cancer | hMLH-1, hMSH-2, hMSH-6, PMS-1, PMS-2 | Colorectal cancer at young age (especially < 50 years); endometrial carcinoma |
| Familial breast cancer | BRCA-1, BRCA-2 | Breast carcinoma at young age (e.g. 20–40 years), may be bilateral; ovarian carcinoma |
| Multiple endocrine neoplasia (MEN) 1 and 2 | MEN | Adenomas within endocrine glands; medullary thyroid carcinoma (in MEN 2) |
| Familial retinoblastoma | RB1 | Retinoblastoma at young age, often bilateral |
| Li-Fraumeni syndrome | p53 | Multiple neoplasms, especially soft tissue sarcomas |
Neoplasms occurring within inherited syndromes usually also occur sporadically (i.e. within individuals who have not inherited a genetic mutation) but with different clinicopathological features. For example, neoplasms occurring within an inherited syndrome usually develop at an earlier age and are more commonly multiple. These conditions may be associated with additional non-neoplastic clinical features e.g. familial adenomatous polyposis (Fig. 3.33.2) is associated with abnormal retinal pigmentation termed Roth spots.
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


