8 Bone tumours and other local conditions
TUMOURS OF BONE
Primary bone tumours, both benign and malignant, are relatively uncommon in comparison with the malignancies arising in other tissues of the body. They are also much less common than metastatic (secondary) tumours which affect the skeleton by blood stream spread from primary carcinoma of the breast, prostate, lung or kidney.
The importance of primary bone tumours is not their frequent occurrence, but the difficulty they may present in diagnosis and treatment and the need to distinguish them from a number of tumour-like lesions that affect bone. Tumours originating in bone arise from mesenchymal tissue and if malignant are termed sarcoma. They are normally classified by the predominant cell type in the lesion, which may be bone, cartilage or fibrous tissue (Table 8.1).
Table 8.1 Classification of primary bone tumours
| Benign | Malignant |
|---|---|
| Arising from bone | |
| Osteoma | Osteosarcoma |
| Osteoid osteoma | |
| Osteoblastoma | |
| Giant-cell tumour | |
| Arising from cartilage | |
| Enchondroma | Chondrosarcoma |
| Osteochondroma (cartilage capped exostosis) | |
| Chondromyxoid fibroma | |
| Chondroblastoma | |
| Arising from fibrous tissue | |
| Fibrous cortical defect | Malignant fibrous histiocytoma (MFH) |
| Non-ossifying fibroma | |
| Fibrous dysplasia | |
| Tumours of uncertain origin | |
| Simple bone cyst | Ewing’s sarcoma |
| Aneursymal bone cyst | Adamantinoma |
Margin of the lesion. Slow-growing tumours have well-defined sometimes sclerotic margins and are most likely benign. Rapidly growing lesions will have ill-defined margins and appear to permeate diffusely into the surrounding bone. While this typically indicates a malignant process, other rapidly progressing conditions, such as acute osteomyelitis, can give a similar appearance.
Breach of cortex. Destruction of the cortex indicates an aggressive invasive lesion.
Biopsy. In the majority of cases it is necessary to undertake a biopsy of material from the lesion for histological and bacteriological examination to reach a final definitive diagnosis. The biopsy may be an open procedure, or a closed needle or trephine technique may be used. The closed technique may be guided by CT scanning, but gives very little material for additional investigations, such as immunohistochemistry or cytogenetics. Needle biopsy should only be used where advice is available from an expert bone pathologist with the necessary technical resources for processing small tissue samples. Once diagnosed the treatment of the lesion depends on whether it is benign or malignant and will be described in more detail for the individual types of tumour.
BENIGN TUMOURS OF BONE
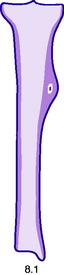
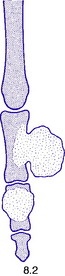
Fig. 8.2 Two types of chondroma: ecchondroma on proximal phalanx; enchondroma in middle phalanx. (See also Figs 6.4A, B)
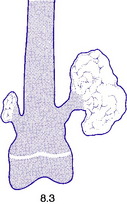
Fig. 8.3 A small and a large osteochondroma. Originating at the growth cartilage, they have migrated away from it with growth of the bone. Each is capped by cartilage. (See also Fig. 6.3B)
Osteoid osteoma
This is a benign circumscribed lesion that may arise in the cortex of long bones or occasionally in the cancellous bone of the spine. It affects young patients aged 10–35 and is three-times commoner in males.
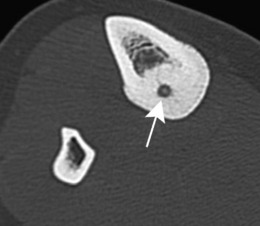
Pathology. The characteristic feature is the formation of a small nidus of osteoid tissue, usually less than 0.5 cm diameter, surrounded by a reactive zone of dense sclerotic new bone formation (Fig. 8.1).
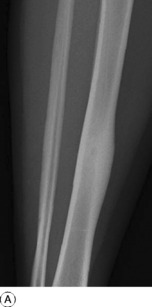
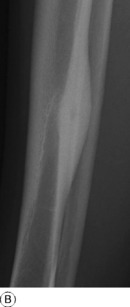
Imaging. Plain radiographs typically show local sclerotic thickening of the shaft that may obscure the small central nidus within the area of rarefaction (Fig. 8.5). The nidus is best seen on a fine cut CT scan (Fig. 8.6) and also exhibits intense uptake on an isotope bone scan.
Chondroma
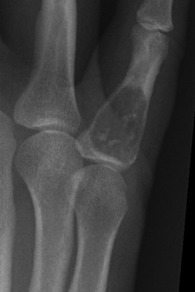
Pathology. There are two forms of chondroma: in the commonest type the tumour grows within a bone and expands it (enchondroma) (Fig. 8.2); in the other more rare form it grows outward from a bone (periosteal chondroma or ecchondroma). Most periosteal chondromas arise in the hands or feet, or from flat bones such as the scapula or ilium. They often reach a large size and are more prone to develop malignant change. Enchondromas are fairly common in the long bones, but over 50% occur in the small bones of the hands and feet. The affected bone is expanded by the tumour and its cortex is much thinned; so pathological fracture is common and usually the presenting feature. Many remain asymptomatic and are only discovered as chance findings on radiographs taken for other purposes.
Imaging. Central enchondromas expand the bone with thinning, but not erosion, of the cortex and exhibit a variable degree of mineralisation or speckled calcification (Fig. 8.7).
Multiple enchondromata of the major long bones occur mainly in the distinct, but rare, clinical condition known as dyschondroplasia (multiple chondromatosis or Ollier’s disease) (p. 65). In this disorder, which begins in childhood, enchondromata arise in the region of the growing epiphysial cartilages (growth plates) of several bones: they interfere with normal growth at the epiphysial plate and consequently may lead to shortening or deformity (see Fig. 6.4A).
Osteochondroma (osteocartilaginous exostosis)
This is the commonest benign tumour of bone, usually presenting in the 10–20 age group.
Pathology. The tumour originates in childhood from the growing epiphysial cartilage plate, but as the bone grows in length the outgrowth gets ‘left behind’ and tends to point away from the adjacent joint. It frequently grows outwards from the bone like a mushroom with a bony stalk in continuity with the cortex of the underlying bone (Fig. 8.3). Less commonly the lesion may be sessile with a more broad-based origin. The bony stalk has a larger cap of cartilage which continues to grow until the cessation of skeletal growth.
The ordinary osteochondroma is single; but in the condition known as diaphysial aclasis (multiple exostoses) (p. 63) the tumours affect several or many bones. The risk of malignant change to a chondrosarcoma is higher in these multiple lesions than in the solitary lesion and should be suspected if the tumour continues to enlarge or becomes painful after puberty.
Imaging. Plain radiographs show the mushroom-like stalk of the bony tumour (Fig. 8.8), but not the larger cartilaginous cap until this calcifies once skeletal maturity is reached. Patients with known lesions should be warned to seek referral for further imaging if their swelling enlarges or becomes painful.
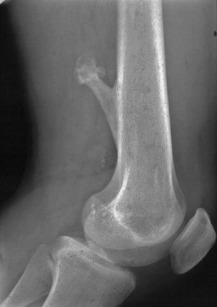
Giant-cell tumour (osteoclastoma)
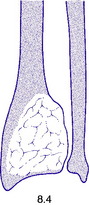
Pathology. The commonest sites are the lower end of the femur, the upper end of the tibia, the lower end of the radius, and the upper end of the humerus – that is, at those ends of the long bones at which most growth occurs. It may also occur in the spine and sacrum. Characteristically it occurs in the end of the bone, occupying the epiphysial region and often extending almost to the joint surface (Fig. 8.4). It destroys the bone substance, but new bone forms beneath the raised periosteum, so that the bone end becomes expanded and pathological fracture is common.
Histologically the tumour consists of abundant mononuclear oval or spindle-shaped stromal cells profusely interspersed with giant cells that may contain as many as fifty nuclei (Fig. 8.9), hence the name ‘giant cell tumour’. The giant cells possibly represent fused conglomerations of the oval or spindle-shaped stromal cells, which may frequently show mitotic figures, though this is not necessarily indicative of malignant change.
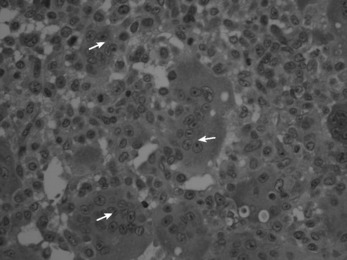
Fig. 8.9 Histology of giant cell tumour showing large osteoclastic multinucleate giant cells (arrows) with interspersed mononuclear tumour cells. The nuclei of the two cell types are very similar. (Haematoxylin and eosin ×400.)
Imaging. Radiographs show lytic destruction of the bone substance, with expansion of the cortex, but no sclerotic rim or periosteal reaction (Fig. 8.10). A few bony trabeculae may remain within the tumour giving a faintly loculated appearance. The tumour tends to grow eccentrically, and often extends as far as the articular surface of the bone. Magnetic resonance imaging will help to determine the amount of soft tissue extension of the tumour (Fig. 8.11).
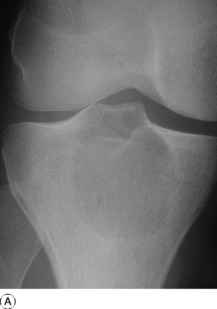
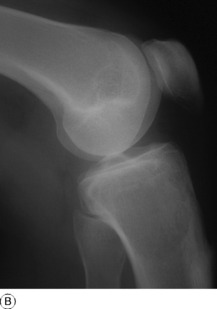
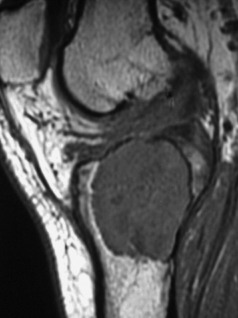
Treatment. This depends upon the site of the tumour. Curettage of the contents with a high-speed burr is the standard method of treatment for most giant cell tumours, though this is associated with a high rate (20–25%) of recurrence. This rate can be reduced to less than 10% by the use of adjuvant treatment applied to the lining of the cavity after curettage. Methods used include the chemical phenol, freezing with liquid nitrogen, or the insertion of polymethylmethacrylate bone cement. In addition to its exothermic reaction on any residual cells, the cement has the added advantage of providing support to the subchondral bone and cartilage of the articular surface of the joint. In some bones, or after multiple recurrences, it may be possible to excise the tumour and replace it with allograft bone or a metal prosthesis, without undue functional compromise. Sites where this is possible include the distal radius, distal femur, and proximal tibia.




