MOLECULAR BIOLOGY OF TUMORS
In the last 30 years, the use of adjuvant chemotherapy has led to dramatic improvement in the survival of children with previously lethal sarcomas. While 30 years ago, 80% of children with a primary bone sarcoma died, now at least that same number will survive (
1,
2). One of the intriguing aspects of childhood sarcomas is that, despite similar histologies, stages, and prognostic factors, some patients respond well to treatment, whereas others seem to be resistant to chemotherapy. To date, patients with good prognoses cannot be distinguished from those with poor prognoses except by crude clinical characteristics, such as the presence of metastatic disease at diagnosis or the histologic response to preoperative chemotherapy (
3). Recent molecular findings in sarcomas may shed light on their biologic behavior and their response to chemotherapy.
One method of looking for genetic alterations in tumors is to examine the chromosomes by karyotype analysis. The identification of recurrent chromosomal abnormalities provides clues regarding sites of potential gene mutations. Normally, there are 23 pairs of chromosomes in the nucleus of the human cell. Osteosarcomas in general have multiple, bizarre karyotypic abnormalities: some chromosomes are missing, some are duplicated, and some are grossly altered. To date, all studies of high-grade osteosarcomas have shown complex karyotypes and nonclonal chromosome aberrations superimposed on complex clonal events (
4,
5). Low-grade juxtacortical osteosarcoma, on the other hand, is characterized by the presence of a ring chromosome accompanied by few other abnormalities or none at all (
6). Although it is usually possible to distinguish high-grade from low-grade osteosarcoma by standard histology, the karyotype information may be diagnostically useful in the case of other tumors. In addition to possibly providing prognostic information, the specific chromosomal aberrations provide clues that assist molecular biologists who are looking for gene mutations (
6).
In contrast to osteosarcoma, Ewing sarcoma (EWS)/peripheral neuroectodermal tumors (PNETs) and alveolar RMSs have single chromosomal translocations characteristic of their respective histologies. In these tumors, part of one chromosome is transposed to part of another chromosome through a breakpoint. A novel gene and gene protein product are created that presumably give the cell a growth advantage. The most common translocations for these tumors are listed in
Table 13-1 (
7,
8).
The demonstration of translocations has been useful in the differential diagnosis of round cell tumors. Under the light microscope, there is little to distinguish one of these tumor types from another, and although immunohistochemistry helps to a certain extent, it is at times difficult to be sure of the diagnosis. Demonstration of these characteristic karyotypic findings makes pathologists more secure in their diagnosis and has helped with the classification of these tumors. To perform a karyotype analysis, short-term cultures and metaphase spreads are necessary, but these are labor- intensive and require fresh tissue (
7). Fluorescent
in situ hybridization and reverse transcriptase-polymerase chain reaction (RT-PCR) allow rapid analysis for the presence of translocations; these techniques can be performed on frozen tissue and sometimes even on paraffin-embedded tissue (
8,
9 and
10). Therefore, it is important to give the pathologist appropriate fresh tissue to be snap frozen to preserve messenger ribonucleic acid (mRNA) and allow these studies to be performed (
11).
These translocations have significance beyond merely establishing the diagnosis. These rearrangements lead to novel proteins that give the tumor cell a growth advantage. In EWS/PNET, for instance, a fragment of the EWS gene contains DNA-binding domains of the FLY1 gene. The protein acts by disrupting pathways that regulate DNA transcription (
12). For several years, it was difficult to make the distinction between EWS and PNET, and clinicians were not sure whether to treat them differently. The observation that both EWS, a poorly differentiated mesenchymal tumor of uncertain cell lineage, and PNET, a tumor believed to be of neural crest origin, shared the same chromosomal translocation led pathologists to believe that both were related neuroectodermal tumors (
13). As noted in
Table 13-1, further studies revealed other translocations in several of these tumors, each such translocation specifying a different novel protein. There is debate regarding whether one or the other of these is associated with a better prognosis, but the treatment strategies used today are the same for both tumors. While some authors suggest that tumors with the type 1 transcript (EWS-FLY1) are associated with a better prognosis than those with other transcripts, others have disputed this (
14,
15).
More recently, these markers have been used in staging and follow-up of high-risk patients (
16). Using RT-PCR technology, one can detect small numbers of tumor cells in a bone marrow or a peripheral blood cell population (
17). This makes the interpretation of bone marrow aspirates more precise and may provide a method for the earlier detection of relapses after treatment. It is hoped that the gene products of these translocations can also be used in treatment strategies. Because the novel genes formed from the translocation make a novel protein that normal cells do not make, antibodies or targeted T cells can be generated to specifically kill tumor cells. This is being tried in early-phase trials of relapsed patients with RMS and EWS/PNET, and if it works, it may be a way of treating patients who fail standard drug therapy.
Genetic alterations in the DNA of sarcomas have been well demonstrated. Mutations in genes, called
oncogenes, give some evidence about the pathogenesis of these tumors and may have some prognostic and therapeutic import (
4,
18,
19). Oncogenes are normal cellular genes (
protooncogenes) that are necessary for the normal development and functioning of the organism (
20). When they are mutated, they may produce a protein that is capable of inducing the neoplastic state. Oncogenes act through a variety of mechanisms to deregulate cell growth. This is obviously a very complex process and may involve more than one genetic event.
There are two categories of oncogenes:
dominant oncogenes and
tumor-suppressor genes (
20). The cumulative effect alters proteins that function as growth factors and their receptors, kinase inhibitors, signal transducers, and transcription factors (
12). The dominant oncogenes encode proteins that are involved in signal transduction, that is, in transmitting an external stimulus from outside the cell to the machinery that controls replication in the cell nucleus. Mutant cellular signal transduction genes keep the cell permanently “turned on.” The protein products of oncogenes also function as aberrant growth factors, growth factor receptors, or nuclear transcription factors. These types of genes seem to have less of a role in osteosarcomas. One exception is amplification of the
HER-2/NEU/ERBB-2 protooncogene in patients with breast cancer, which confers a poorer prognosis. Patients with this amplification are treated with a monoclonal antibody to this protooncogene [MAb45D5, trastuzumab (Herceptin)]. Overexpression of
HER2-NEU in osteosarcoma has been reported and is associated with advanced disease and poorer prognosis (
21,
22). Although this has been disputed by some studies (
23,
24), it provides the potential for treatment strategies in patients with osteosarcoma who have amplification of HER2-NEU.
A second class of genes are the tumor-suppressor genes, which encode proteins whose normal role is to restrict cell
proliferation (
25,
26). They act as brakes rather than as accelerators of growth. Their normal role is to regulate the cell cycle and keep it in check. The retinoblastoma gene (
RB) was the first gene recognized in this class (
27). Osteosarcomas are very frequent in patients with hereditary retinoblastoma (1000× increased chance), both in the orbit and in the extremities, and are unrelated to irradiation. It was subsequently learned that osteosarcoma in these patients, as well as spontaneously occurring osteosarcomas, carries mutations or deletions of the
RB gene. It was one of the first clues to the finding that osteosarcomas have a genetic cause. It is estimated that approximately 60% to 75% of sporadic osteosarcomas either have an abnormality of the
RB gene or do not express a functional RB product (
19). The
RB gene is located on the long arm of chromosome 13 (13q14) and is 200 kb in length. Its product is a 105- to 110-kDa nuclear phosphoprotein (pRB) that appears to have a cell cycle regulatory role. The retinoblastoma protein acts as a signal protein, or a gatekeeper, to regulate the cell cycle through the transcription of genes that mediate the cell cycle. Deactivation of the
RB gene or absence of pRB allows cells to enter the cell cycle in an unregulated fashion, a condition that imparts a growth advantage to the affected cell. It should be noted that one copy of the gene is sufficient for a normal phenotype. A child born with a normal allele and a mutant or an absent allele will not manifest retinoblastoma until some event occurs in retinoblasts to alter the normal allele. If both copies become deranged, the normal check on the cell cycle disappears, and the conditions for the neoplastic state are met. There are several other mechanisms by which the function of the RB protein can be altered; for instance, viral proteins may bind to the RB protein and inactivate it (
5).
The second tumor-suppressor gene to be identified was the
p53 gene (
28,
29 and
30). Located on the short arm of chromosome 17 (17p), its product is a nuclear phosphoprotein that has a cell cycle-regulatory role similar to that of the RB protein. As in the case of
RB, inactivation of p53 gives the cell a growth advantage, probably because of loss of cell cycle regulation. The p53 phosphoprotein may be inactivated by a variety of mutations, including a single base change (point mutation) that increases the half-life of the protein, allelic loss, rearrangements, and deletions of the
p53 gene. Each of these mechanisms can result in tumor formation by loss of growth control. The p53 protein functions as an extremely important cell cycle checkpoint that blocks cells with DNA damage until they can be repaired or directs damaged cells into apoptosis (programmed cell death) if they cannot be repaired. Cells lacking this checkpoint can accumulate successive genetic abnormalities and possibly become malignant. It is estimated that approximately 25% of osteosarcomas have detectable mutations of the
p53 gene (
31).
The p53 protein is a transcription factor, meaning that it binds to regions of other genes (DNA) and controls the expression of genes responsible for cell cycle control (cell growth), apoptosis (programmed cell death), and other metabolic functions, such as control and repair of DNA damage. In concert with RB and a variety of other proteins, p53 acts to regulate the cell cycle through a complex cascade of enzymes, in which RB probably plays the central role. Apoptosis has recently become recognized as an important mechanism by which chemotherapy and radiotherapy kill cancer cells. p53 is involved in this process and appears to arrest cell division after sublethal damage (e.g., by radiation), to give the cell time to repair DNA defects before the next division (
32,
33 and
34). If repair does not take place, the cell undergoes apoptosis and dies. If p53 is not functional, the cell may survive and accumulate genetic defects, leading to malignant transformation. Osteosarcomas have been shown to have a variety of mutations of the
p53 gene (
35,
36 and
37). Preliminary evidence suggests that overexpression of mutant p53 protein (detected by immunohistochemistry) or loss of heterozygosity of the
p53 gene is related to human osteosarcoma (
38,
39).
In sarcomas, genetic defects other than p53 and RB have also been detected. One example is a gene called
mdm-2, which is a zinc finger protein that is amplified in some sarcomas (
28,
40,
41). It inactivates p53 protein by binding to it, preventing its transcription factor activity. Cordon-Cardo et al. (
42) studied 211 adult STSs by immunohistochemistry, using monoclonal antibodies to mdm-2 and p53, and demonstrated a correlation between overexpression of mdm-2/p53 and poor survival rates. Patients without mutations in either gene (mdm-2/p53-) had the best survival rates, those with one mutation (either mdm-2+/p53- or mdm-2-/p53+) had intermediate rates of survival, and those with mutations in both genes (mdm-2+/p53+) had the lowest survival rates. Another mechanism in which p53 protein can be inactivated is by viral proteins that bind and inactivate both RB and p53 protein (
43).
Not only are genetic mutations found in the tumors of patients with sarcomas, but mutations may also be present in all somatic cells (
germ-line mutations) in patients with heritable cancer (
44,
45 and
46). Although such defects do not appear to be common in the general population, germ-line p53 mutations are present in patients who are part of a familial cancer syndrome. These families have a variety of cancers, often at an early age, and osteosarcomas and STSs are a fairly common occurrence in these kindred. Identification of patients with p53 germ-line mutations can be useful in determining which patients in an affected family are at risk for developing cancers, but much more work is needed in the area of genetic counseling to determine how best to use this information. One study showed that germ-line mutations were present in approximately 3% to 4% of children with osteosarcoma, and that the detection of these mutations was more accurate than family history in predicting the family’s susceptibility to cancer (
47).
How is this information useful for treatment? One possibility is that the p53 mutations may be potential biologic markers of prognosis and response to treatment (chemotherapy). There is some preliminary evidence that p53 mutations in the tumor may portend a worse prognosis in osteosarcoma. More recently, the association of p53 with apoptosis has suggested possible strategies for chemotherapy, on the basis of the status of the p53 pathway (
33,
34). Gene therapy (replacing the missing or mutated gene by transfection with viral carriers) is often discussed, but there are major technical hurdles to overcome before this technology can be used for treating cancers
in humans. However, it might be possible to make tumor cells more antigenic, or to make them more sensitive to antineoplastic drugs, by gene transfer. Another strategy would be to alter normal cells to make them less sensitive to damage by chemotherapeutic agents. Currently, these techniques pose technical challenges, but they offer realistic promise for the near future.
Another exciting area of research in the molecular biology of sarcomas is multidrug resistance (MDR). MDR probably explains why some patients respond to chemotherapy and others do not. Drug resistance may be intrinsic (present at diagnosis) or acquired (appearing after treatment of a tumor) (
48,
49). At least four basic mechanisms of drug resistance are now recognized under the category of the MDR phenotype. They are (a) changes in glutathione metabolism, (b) alterations in topoi-somerase II, (c) non-P-glycoprotein (P-gp)-mediated mechanisms, and (d) P-gp-mediated mechanisms (
6,
7,
48,
49 and
50). Recent evidence has suggested that P-gp may be of particular relevance to osteosarcoma. P-gp is a glycoprotein encoded by the
MDR-1 gene on the long arm of chromosome 7 in humans (
48,
49).
MDR-1 is one member of the aneurysmal bone cyst (ABC) superfamily of genes that encode membrane transport proteins; these proteins function as unidirectional membrane pumps using adenosine triphosphate hydrolysis to work against a concentration gradient. P-gp is a 170-kDa protein that is located in the cell membrane and functions as an energy (adenosine triphosphate)-requiring pump that excludes certain classes (amphipathic compounds) of drugs from the cell. This physiologic mechanism is believed to be important in certain organ systems, such as the blood-brain barrier, placenta, liver, kidney, and colon, for ridding the cell of unwanted toxins, but it is also responsible for actively excluding chemotherapeutic agents, such as
Vinca alkaloids, anthracyclines, colchicine, etoposides, and taxol (many of which are active in osteosarcoma protocols) from the cancer cell. Another feature of the P-gp mechanism that may have some relevance to therapeutic strategies is that some classes of drugs can reverse the MDR phenotype by blocking the action of the pump. These drugs include verapamil, cyclosporin A, tamoxifen, and others.
Several studies have demonstrated that some sarcomas (25% to 69%) display the MDR phenotype at diagnosis, and that relapsed sarcomas show higher incidence and intensity of MDR expression (
48,
49,
51,
52). Because of the small numbers of patients in these studies, and the variety of the methods by which MDR expression was tested, comparisons of the studies and an accurate determination of the incidence of MDR expression are difficult to accomplish. In addition, the age of the patient and the type of sarcoma appear to be related to the incidence of detectable P-gp at diagnosis. One study showed that osteosarcomas have a higher incidence of MDR than other types of adult sarcomas (
51). Serra et al. (
53) demonstrated that overexpression of P-gp protein was evident in 23% of primary and 50% of metastatic osteosarcomas.
Baldini et al. (
54) reported on 92 patients with non-metastatic osteosarcoma of an extremity who had been treated with chemotherapy and surgery. The study demonstrated that an immunohistochemically determined expression of P-gp predicted a decreased probability of the patient having an event-free survival, and was more accurate in prediction than histologic response to preoperative chemotherapy. Another study failed to find a relation between MDR-1 mRNA expression and outcome in patients treated for osteosarcoma (
55).
Findings such as these are important in planning future protocols in human osteosarcoma. The drug-resistant tumor is becoming better identified as one that has a poor histologic response to preoperative chemotherapy and that expresses P-gp. Undoubtedly, it is more complex than this, and other mechanisms will pertain. Several caveats exist. One is the complexity of defining the resistant tumor. Preoperative chemotherapy requires 10 to 12 weeks to provide an estimate of histologic necrosis, unless ways can be found to accurately predict percentage of necrosis by positron emission tomographic (PET) scans, thallium scans, and/or gadolinium-enhanced magnetic resonance imaging (MRI). Detection of P-gp at diagnosis is difficult, and no one method has proven superior. It is probably not sufficient to demonstrate the presence of P-gp; also important is whether the pump is functioning to exclude cytotoxic agents from the tumor cell. Ideally, one would like to reverse the action of the P-gp mechanism but, just as there are no new agents to rescue patients who show poor histologic response, the agents currently available to reverse MDR are of limited benefit. They are potentially problematic in that they make normal cells less tolerant of chemotherapy, and thereby increase toxicity; and in other tumors they have not proven to be effective. The future probably lies in developing more effective reversing agents and in defining other drug-resistant mechanisms.
EVALUATION
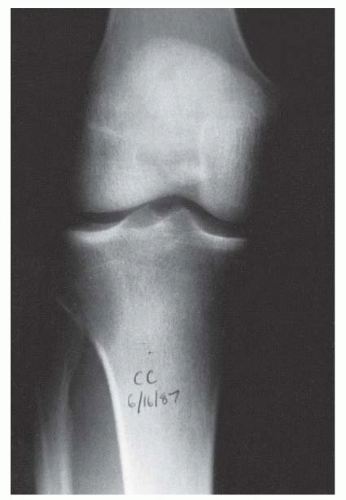
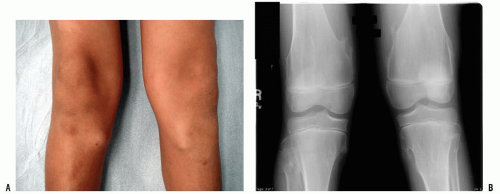
A thorough evaluation is necessary for any child presenting with a bone or soft-tissue mass. Although infection and trauma are much more common than a neoplastic process, the consequences of the mismanagement of a patient with a musculoskeletal tumor can be grave (
Fig. 13-1).
Medical History.
Most children have no significant past medical history, but inquiries should be made. Has the child had a previous fracture? Has the child had other illnesses? Have radiographs been taken previously? Do not assume that the patient or the family will volunteer significant past medical history. Questions that should be asked include: How long has the mass been present? The longer the mass has been present, the more likelihood of a benign process. Especially worrisome are new masses that arise and grow over a short period of time. Is the mass getting bigger or is it stable in size? Masses that are rapidly growing indicate an active process that could be aggressive. Depending on the location, however, such as axial skeleton, some masses may not be noticed until they reach substantial size. Among younger patients, a parent usually notices the mass first, and although the parent will usually think that the mass has appeared overnight, this is rarely the
case. Teenagers may report the presence of a mass, but often only after a few weeks or months of waiting for it to resolve spontaneously. Is the mass associated with pain? Pain at the lesional site is a frequent complaint (see below). Active and aggressive tumors will usually present with pain. Painful soft-tissue masses are most often abscesses. Most soft-tissue tumors do not produce significant symptoms until they are large. Although most of the soft-tissue masses seen in children prove to be benign, all soft-tissue masses, even those in children, should be considered to be malignant tumors until proven otherwise. The consequences of mistaking a malignant soft-tissue tumor for a benign tumor can be devastating, whereas the consequences of approaching a benign tumor as if it were a malignancy are minimal. Is there a history of cancer? Depending on the age and the type of tumor, metastatic disease may be the main differential diagnosis.
Chief Complaint.
Generally, bone and soft-tissue tumors present in one of four ways:
Pain is the most common presenting complaint of a child with a musculoskeletal tumor. The characteristics of the pain can help determine the diagnosis. Ask the patient: Where is the pain? How did it begin? Is it sharp, dull, radiating, or constant? Is it associated with activity? Is there a particular activity that makes the pain worse? What makes the pain better? Does it awaken you at night? Is the intensity of the pain increasing, staying the same, or diminishing?
Patients who have active or aggressive benign tumors (e.g., ABC, chondroblastoma, and osteoblastoma) usually have a mild, dull, slowly progressive pain that is worse at night and aggravated by activity. Patients with malignant musculoskeletal tumors complain of a more rapidly progressive symptom complex, not specifically related to activity, which often awakens them at night. Occasionally, the pain pattern is diagnostic. The classic example is the pain of an osteoid osteoma, which is a constant, intense pain that is worse at night, and is almost always relieved by aspirin or nonsteroidal anti-inflammatory drugs (NSAIDs). The pain caused by a Brodie abscess (subacute osteomyelitis) is similar to that of an osteoid osteoma, but is rarely relieved by aspirin.
Most children and parents date the onset of symptoms to a traumatic event. The specific nature of the trauma and the relation of the trauma to the current symptoms must be evaluated thoroughly. Trauma without a definitive fracture may be the explanation for an abnormal radiograph, but it should not be assumed to be the explanation, even for a periosteal reaction, unless the history is perfectly consistent. With the increased level of organized sports for children, there has been an increase in the incidence of fatigue or stress fractures, and these can sometimes be confused with neoplasias. Still, one should be cautious about ascribing a lesion to trauma.
The child presenting with a fracture should be questioned about the specifics of the injury that produced the fracture. Most lesions that lead to a pathologic fracture are easily recognized on a plain radiograph, but occasionally they may not be obvious. When the traumatic event seems insignificant, a pathologic fracture should be suspected. Patients should be asked about symptoms, no matter how minimal, that they experienced before the fracture. Most aggressive benign tumors and malignant tumors produce pain before the bone is weakened enough to fracture. Latent benign tumors such as unicameral bone cyst (UBC) and nonossifying fibroma (NOF) are often diagnosed following a trauma, as an incidental finding or a pathologic fracture.
A complete review of systems is mandatory. Ask specifically about fever, decreased appetite, irritability, and decreased activity. Most patients with musculoskeletal tumors do not have systemic symptoms at presentation, and their presence should alert the physician to the possibility of an underlying generalized disorder or osteomyelitis. Rarely, children with a malignant neoplasm, such as EWS, may present with fever, weight loss, and malaise, favoring an infectious etiology. Even
children with large primary malignant musculoskeletal tumors usually appear healthy.
Physical Examination.
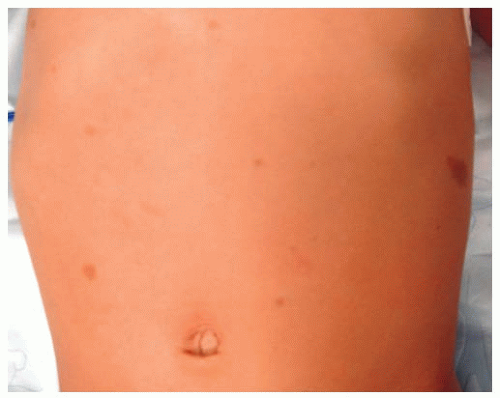
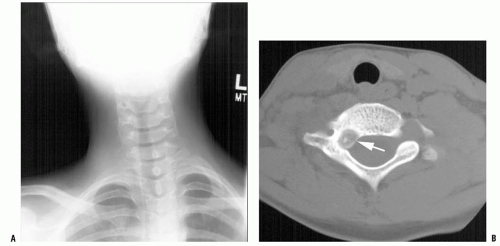
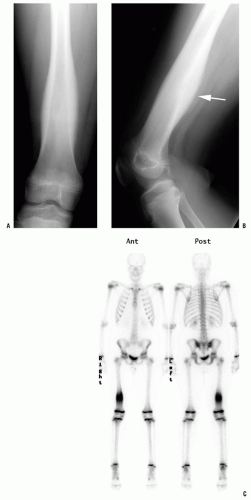
All patients with musculoskeletal complaints, especially those in the pediatric age group, should have a complete physical examination. Not only can important information be gained about the specific disorder being evaluated, but also other significant abnormalities may be found. For example, café au lait lesions of the skin are a clue that the patient has fibrous dysplasia or neurofibromatosis (
Fig. 13-2); numerous hard, nontender, fixed masses near the ends of long bones are suggestive of multiple hereditary exostosis (MHE).
The affected extremity should be examined carefully. The mass should be measured; larger tumors are usually more active and worrisome. Although there isn’t a specific number, soft-tissue masses over 5 cm and bone tumors over 8 cm have a higher likelihood of being malignant. The location is an important characteristic. STSs are usually located deep to the deep fascia, while bone sarcomas are usually located around the fastest growth areas (e.g., knee and shoulder). STSs are usually “fixed” to superficial or deep structures (no mobility) and firm to touch; soft, movable, nontender masses, especially those in the subcutaneous tissues, are usually benign. Transilluminate the mass, if light is transmitted more easily through the mass than through the surrounding tissue, the mass is a fluid-filled cyst. The gait pattern should be recorded; muscular atrophy measured, and the range of motion of the adjacent joint should be measured. The presence of erythema, tenderness, or increased temperature should be noted.
Neurovascular exam is essential. Often vascular malformations will be in the differential of soft-tissue tumors; check for pulsations or bruit. Detailed peripheral nerve check will assist in evaluating the proximity to these structures. Check for satellite lesions, the easiest lesion to miss is the second lesion. Examine the abdomen for hepatomegaly, splenomegaly, etc. Examine regional lymph nodes; although most musculoskeletal malignancies metastasize via hematogenous, some will do it via lymphatic. The most common ones are epithelioid sarcoma (16%), synovial sarcoma (15%), RMS (13%), and angiosarcoma (13%) (
56).
Plain Radiograph Examination.
Plain radiographs are the single most useful image modality to assess a musculoskeletal tumor; all patients should have at least anteroposterior and lateral plain radiographs of the affected area. Often bone tumors are incidentally found after radiographs are taken for other reason. Pathologic fracture is also a common presentation, especially among some benign tumors such as UBC.
The entire lesion must be observed. The radiograph should be reviewed systematically. Look at the bone, all of it, and every bone on the radiograph. Ask yourself these questions: Is there an area of increased or decreased density? Is there endosteal or periosteal reaction, and if there is, what are the characteristics of the reaction? Is there cortical destruction? Is it localized or are there multiple defects? Is the margin in the tumor well defined or poorly defined? Is there a reactive rim of bone surrounding the lesion? Are there densities within a radiolucent lesion? Is the bone of normal, increased, or decreased overall density? Is the joint normal? Is there loss of articular cartilage? Is the subchondral bone normal, thick, or thin? Are there abnormalities in the bone on both sides of the joint? Are there intra-articular densities? Is there a soft-tissue mass? Are there calcifications or ossifications in the soft tissue? If one looks specifically for abnormalities, it is unlikely that an abnormality will be missed.
The pelvis and the scapula are exceptions to this rule. Large tumors involving the pelvis or the scapula, even those with marked destruction of bone, can be extremely difficult or impossible to see on a plain radiograph. If there is a suggestion that the patient has a pelvic or a scapular tumor, computerized axial tomography (CT) scan or magnetic resonance (MRI) is recommended.
Enneking (
57) proposes that four sets of questions should be asked when looking at plain radiographs of a possible bone tumor.
Where is the tumor? This refers to the lesion’s anatomic location: long bone or flat bone; epiphyseal, metaphyseal, or diaphyseal; and medullary canal, intracortical, or surface. Based on the tumor location and the patient’s age, one can already formulate a differential list.
What is the tumor doing to the bone? Is there erosion of the bone, and if so, what is the pattern? This will determine the lesion aggressiveness.
What is the bone doing to the tumor? Is there periosteal or endosteal reaction? Is it continuous? Is it sharply defined? The periosteal reaction will reflect the efforts of the host bone to contain the lesion.
Are there any intrinsic characteristics within the tumor that indicate its histology? Is there bone formation by the tumor? Is there calcification? Is the lesion completely radiolucent?
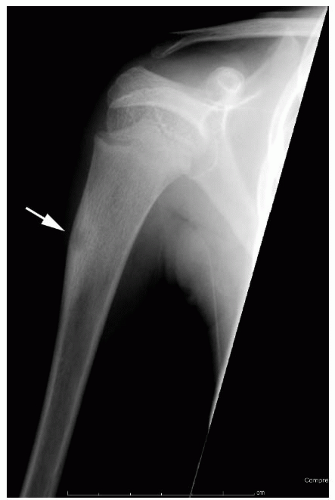
In addition to this list approach, always consider patient’s age and specific location of the tumor within the bone, as these characteristics will limit the differential diagnosis (
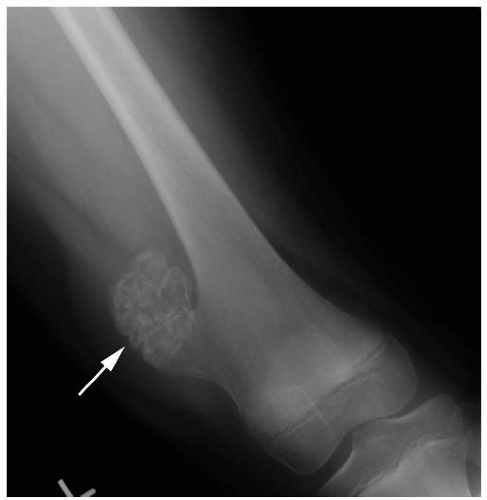
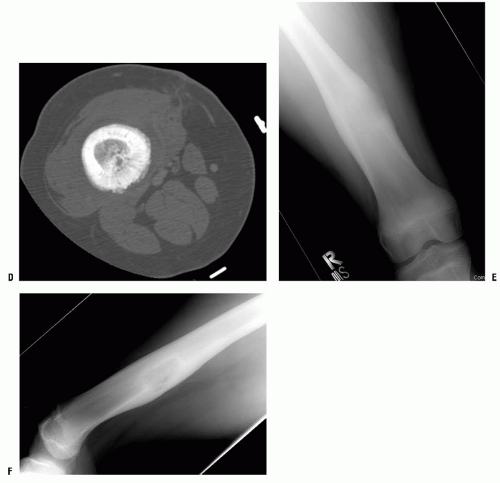
Table 13-2). Most bone tumors can be diagnosed correctly after obtaining the history, performing a physical examination, and examining the plain radiograph. When the specific diagnosis is made from these examinations, additional studies are requested only if they are necessary for treatment. Often, specific treatment can be planned from only the history, physical examination, and plain radiographs. For example, a 12-year-old boy with a hard, fixed mass in the distal femur that has been present for several years and has not increased in size for more than 1 year complains of pain after direct trauma to this mass. Plain radiographs confirm the clinically suspected diagnosis of osteochondroma (
Fig. 13-3). Further evaluation to make the diagnosis is not necessary.
When the specific diagnosis cannot be made, it should be possible to limit the differential to three or four diagnoses, and appropriate additional evaluations can be requested. CT, MRI, and nuclear bone scanning (technetium, gallium, thallium, or indium) may reveal findings that are diagnostic, or that provide the information required for planning a subsequent biopsy.
Additional Diagnostic Studies
Laboratory Examinations.
For the most part, serum and urine laboratory values are usually normal in musculoskeletal neoplasia. Nonetheless, a few musculoskeletal tumors are associated with abnormal laboratory values. The erythrocyte sedimentation rate (ESR) is nonspecific but sensitive. Patients with infections or malignant tumors usually have an elevated ESR, but patients with benign disease should have a normal value. A normal ESR value can increase the physician’s confidence that a suspected benign, inactive lesion is just that. Patients with active benign or malignant musculoskeletal tumors, particularly those with EWS, often have an elevated ESR, but it is rarely >80 mm/hour. A markedly elevated value (>180 mm/hour) favors a diagnosis of infection and may be just what is needed to justify an early aspiration of a bone or soft-tissue lesion. C-reactive protein (CRP) is another useful serum value that indicates systemic inflammation. Because it increases and returns to normal more quickly than ESR, CRP has been used as the main serum value to follow-up infection.
Serum alkaline phosphatase is present in most tissues in the body, but the bones and the hepatobiliary system are the predominant sources. In the pediatric age group, conventional high-grade osteosarcoma is associated with
elevated levels of serum alkaline phosphatase (
58). Not all patients with osteosarcoma have elevated levels of serum alkaline phosphatase, and therefore a normal level does not exclude osteosarcoma from the diagnosis. A minimal elevation can be observed with numerous processes, even a healing fracture. Adults with elevated levels of serum alkaline phosphatase secondary to bone disease are most likely to have Paget disease of bone or diffuse metastatic carcinoma. Patients with a primary liver disorder have elevated levels of serum alkaline phosphatase as well, but they also have elevated levels of serum 5-nucleotidase and leucine aminopeptidase, and glutamyl transpeptidase deficiency. The levels of 5-nucleotidase and leucine aminopeptidase are not elevated in primary bone tumors. Two- to threefold increase in the alkaline phosphatase levels has been associated with worse prognosis in patients with osteosarcoma (
58).
Serum and urine calcium and phosphorus levels should be measured, especially if a metabolic bone disorder is suspected. Serum lactate dehydrogenase (LDH) level is elevated in some patients with osteosarcoma. Patients with EWS or osteosarcoma with elevated LDH have a worse prognosis (
15,
59,
60). Elevated LDH levels may also indicate relapse in a patient who has been treated for these tumors (
59). Patients entering chemotherapy treatment protocols will need to have LDH levels determined in order to stratify them on the protocol. Other laboratory determinations are not helpful and are not recommended.
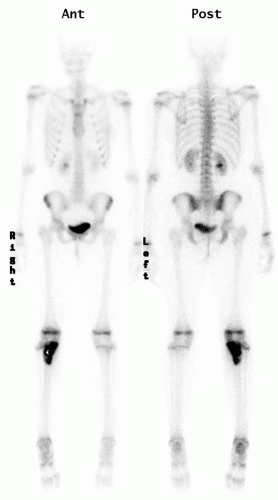
Radionuclide Scans.
Technetium bone scanning is readily available, safe, and an excellent method for evaluating the activity of the primary lesion. In addition, bone scanning is the most practical method of surveying the entire skeleton (
Fig. 13-4). Technetium-99 attached to a polyphosphate is injected intravenously, and, after a delay of 2 to 4 hours, the polyphosphate, with its attached technetium, concentrates in the skeleton proportional to the production of new bone. A disorder that is associated with an increase in bone production increases the local concentration of technetium-99 and produces a “hot spot” on the scan. The technetium bone scan can be used to evaluate the activity of a primary lesion, to search for other bone lesions, and to indicate extension of a lesion beyond what is seen on the plain radiograph. The polyphosphate-technetium-99 compound also concentrates in areas of increased blood flow, and soft-tissue tumors usually have increased activity compared with normal soft tissues. The technetium-99 bone scan can be used to evaluate blood flow if images are obtained during the early phases immediately after injection of the technetium-99. The polyphosphate-technetium-99 is cleared and excreted by the kidneys, so the kidneys and the bladder have more activity than other organs. The technetium-99 scan is sensitive but nonspecific, whereas infectious processes will usually present with “hot scans.” The principal value of a radionuclide scan is as a means of surveying the entire skeleton for clinically unsuspected lesions. There are exceptions and false negative may occur, in approximately 25% of cases of Langerhans cell histiocytosis (LCH), the bone scan is normal, or there is decreased activity at the site of the lesion (
42,
61,
62).
PET is being used more frequently in the evaluation of musculoskeletal tumors (
42,
63). Fluoro-2-deoxy-D-glucose (FDG) PET is the type of PET used most frequently for the musculoskeletal system. Because there is a differential uptake of FDG between neoplastic tissue and normal tissue (neoplastic tissue has greater uptake), it is possible to identify neoplastic tissue on a PET scan. The role of PET in the evaluation and monitoring of patients with musculoskeletal neoplasia is under
investigation, especially among children. PET with fluorine-18-FDG has proved particularly useful in evaluating patients with lymphoma (
64,
65).
Computerized Axial Tomography.
When introduced in the late 1970s, CT scan dramatically improved the evaluation of bone and soft-tissue tumors. The anatomic location and extent of the tumor could be determined accurately. The improved accuracy of anatomic localization means that less radical surgery can be performed safely.
The density of a bone or soft-tissue mass on a CT scan is called its “attenuation coefficient” and is measured in Hounsfield units (HU). The density of water is 0 HU; tissues more dense than water have a positive value, and tissues less dense than water have a negative value. The vascularity of a lesion can be evaluated by measuring the increase in the attenuation coefficient of a lesion after intravenous infusion of contrast, and comparing this increase to that in an adjacent muscle. Normal muscle has an attenuation coefficient of approximately 60 HU, and increases 5 to 10 HU with a bolus of intravenous contrast. Fat has an attenuation coefficient of approximately 60 HU, and cortical bone usually has a value of more than 1000 HU.
CT scan can be performed quickly and is less anxiety producing than closed MR, so sedation is less likely to be needed when compared with MRI. The main downside is the amount of radiation delivered in a CT scan, particularly among children (
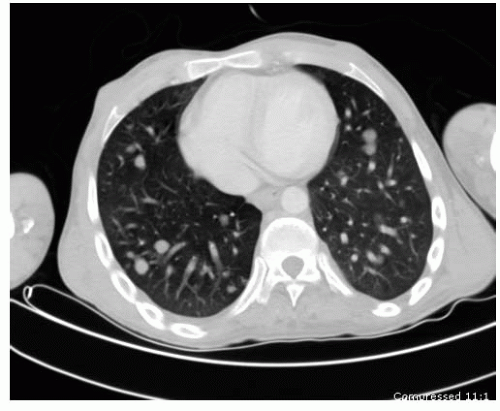
66). CT scan is most useful in the evaluation of small lesions in or immediately adjacent to the cortex (e.g., osteoid osteoma) and lesions with fine mineralization or calcifications (e.g., chondroblastoma). CT is still the gold standard for chest evaluation and to rule out lung nodules (
Fig. 13-5). CT has also been used for percutaneous biopsies and treatment of several different lesions.
Magnetic Resonance Imaging.
MRI does not expose the patient to radiation and has proved to be the most useful tool in the evaluation of soft-tissue lesions. MRI produces images of the body in all three planes (axial, sagittal, and coronal) as easily as in a single plane, and poses no known hazards to the patient.
The images are produced by a computer program that converts the reactions of tissue hydrogen ions in a strong magnetic field excited by radio waves. By adjusting excitation variables, images that are T1- and T2-weighted are obtained. A variety of techniques have been used to produce images of improved quality compared with routine T1- and T2-weighted images. The use of gadolinium as an intravascular contrast agent allows one to judge the vascularity of a lesion, thereby providing even more information about the tumor. Fat-suppression images with gadolinium enhancement are often especially useful in demonstrating a soft-tissue neoplasia. As with CT scan, it is important for the orthopaedist requesting MRI to discuss the case with the radiologist. The radiologist can then determine the optimal MRI settings required for visualizing the lesion.
MRI is the single most important diagnostic test after physical examination and plain radiography for evaluating a musculoskeletal lesion. The ability to view the lesion in three planes, determine its intraosseous extent, see the soft-tissue component clearly, and have an idea of the tissue type from one diagnostic test makes MRI a powerful tool. Unfortunately, variations in technique mean that it is important that the examination be planned carefully if the maximum information possible is to be obtained. T1-weighted (with and without gadolinium), T2-weighted, and fat-suppression techniques are the minimal images needed.
Staging.
Patients with neoplasia can be separated into groups on the basis of the extent of their tumor and its potential or presence for metastasis. These groups are called
stages. Grouping patients by their stage helps the physician predict a patient’s risk of local recurrence, metastasis, and outcome. This facilitates making treatment decisions about individual patients and helps in the comparison of treatment protocols. Staging systems are based on the histologic grade of the tumor, its size and location, and the presence of regional or distant metastases. The presence of a metastasis at the time of presentation is a bad prognostic sign and, regardless of other findings, puts the patient in the highest-risk stage. For patients without metastases at presentation, the histologic grade of the tumor is the principal prognostic predictor. Size is next in importance. Higher histologic grade and larger tumors are associated with the worse prognoses (
67).
There are two common staging systems in use for musculoskeletal tumors. The task force on malignant bone tumors of the American Joint Commission on Cancer Staging and End Result Studies published a staging system for soft-tissue tumors in 1977, which was most recently revised in 2002 (
68). This staging system is based on the histologic grade (G), local extent or size (T), whether the nodes are involved (N), and
metastases (M). The tumors are separated into three histologic grades (G1, low grade; G2, medium grade; G3, high grade) and two sizes (T1 for <8 cm (for bone) or 5 cm (for soft tissue), T2 for equal to or greater than that). Patients with nodal involvement are designated N1, and those without nodal involvement are designated N0. Patients with metastatic disease are designated M1, and those without metastatic disease are designated M0. There are four stages, with subclasses in each stage. Tumors at stage I are associated with the best prognosis, and tumors at stage IV with the worst prognosis.
Enneking et al. (
69) also proposed a musculoskeletal staging system. This system is used more often by orthopaedists involved in the management of patients with musculoskeletal tumors. It was designed to be simple, straightforward, and clinically practical. The tumors are separated into only two histologic grades (I, low grade; II, high grade) and two anatomic extents (A, intracompartmental; B, extracompartmental). Patients with metastatic disease in either a regional lymph node or a distant site are grouped together as stage III. Each bone is defined as its own separate anatomic compartment. The soft-tissue anatomic compartments are defined as muscle groups separated by fascial boundaries. There are five stages in this system (
Table 13-3).
Enneking et al. (
69) also introduced four terms to indicate the surgical margin of a tumor resection. These terms are in common use, and provide a means of describing the relation between the histologic extent of the tumor and the resection margin. The surgical margins are defined as
intralesional, marginal, wide, and
radical. An intralesional margin is the surgical margin achieved when a tumor’s pseudocapsule is violated and gross tumor is removed from within the pseudocapsule. An incisional biopsy and curettage are two common examples of an intralesional margin. A marginal surgical margin is achieved when a tumor is removed by dissecting between the normal tissue and the tumor’s pseudocapsule. This is a surgical margin obtained when a tumor is “shelled out.” A wide surgical margin is achieved when the tumor is removed with a surrounding cuff of normal, uninvolved tissue. This is often referred to as
en bloc resection and is the most common type of resection used for malignant tumors. A radical surgical margin is achieved when the tumor and the entire compartment (or compartments) are removed together. This usually is accomplished only with an amputation proximal to the joint that is just proximal to the lesion (e.g., an above-knee amputation for a tibial tumor). As a rule, benign lesions can be managed with an intralesional or a marginal surgical margin, but malignant tumors require a wide surgical margin. Radical surgical margins are reserved for recurrent tumors and the most infiltrative malignancies.
Biopsy.
Biopsy is an essential part of tumor staging and management decision making for children with a bone or a soft-tissue tumor. Sometimes, a biopsy can be avoided and diagnosis made on basis of history, physical examination, and imaging studies. When a biopsy is required, the prebiopsy evaluation improves the chance that adequate and representative tissue will be obtained, the least amount of normal tissue will be contaminated, and the pathologist will make an accurate diagnosis. It is recommended that the surgeon consult with the radiologist and the pathologist before performing the biopsy to get their suggestions for the best tissue to obtain; furthermore, discussing the case preoperatively with the pathologist will allow the pathologist to be better prepared to make a diagnosis from a frozen section.
The purpose of the biopsy is to confirm the diagnosis suspected by the physician after the evaluation, or to determine which diagnosis, from among a limited differential diagnosis, is correct. In addition to providing confirmation for a specific diagnosis, the tissue obtained must be sufficient for histologic grading. It must be representative of the tumor and, because many musculoskeletal tumors are heterogeneous, the specific site from which the tissue is taken is important. Biopsy is not a simple procedure; the musculoskeletal tumor society has shown that an unplanned or erroneous biopsy can impact negatively the outcome, with higher incidence of unneeded surgery, including amputation and worse outcome (
70).
There are two forms of biopsies: percutaneous (needle biopsy) and open (incisional and excisional). Percutaneous biopsy can be done via fine needle aspirate or core. It has the advantage of having low morbidity and sometimes can be done in clinic (older patients). Some of the disadvantages include a small amount of tissue and a higher chance for sampling error that may limit the ability to perform special stains and cytogenetics. The reported accuracy of a needle biopsy is around 85% (
71).
Open biopsy has the advantage of obtainment of a larger tumor sample that allows the pathologist to perform all necessary studies and decreases the chance of sampling error. The accuracy of open biopsy is close to 96% (
71). Furthermore, most children will require general anesthesia for a biopsy and therefore is important to obtain adequate sampling. Open incisional biopsy is the most commonly used technique. It entails obtaining a sizeable fragment of the tumor without attempting excision of the whole mass. Ideally, the treating surgeon will be the one performing the biopsy. That should avoid several possible complications that could impact in the ability of performing limb salvage and adequate tumor resection. Some of
the principles of open biopsy include drawing definitive limb salvage incision prior to start; avoiding transverse incisions on extremities; avoiding raising flaps or exposure of neurovascular structures; always performing an intraoperative frozen section to ensure acquisition of diagnostic tissue; if a drain is used, it should exit the skin in line with the incision; placing sutures within 5 mm of the incision; sending material for culture and sensitivity; achieving meticulous homeostasis (hematoma from the biopsy may contain tumor cells and will require resection if surgery is the treatment); and avoiding or judicious use of local anesthesia (
72).
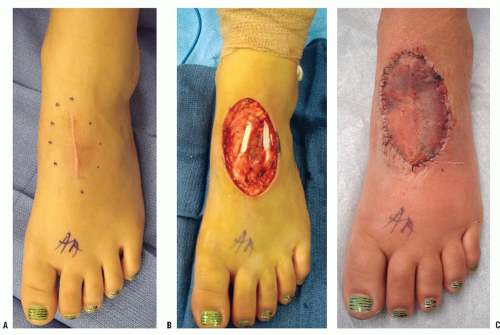
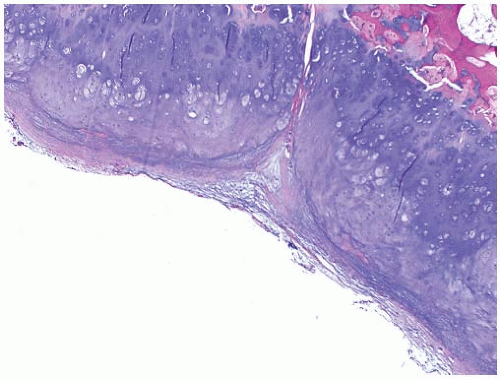
Occasionally an excisional biopsy, rather than an incisional biopsy, is indicated. Open excisional biopsy differs from incisional biopsy in that the entire tumor is excised and sent for analysis. An excisional biopsy is appropriate when the lesion is small and can be excised with a cuff of normal tissue. It is usually reserved for small (<3 cm) lesions that are likely benign. An excisional biopsy may be appropriate even when a major resection is required. If the preoperative evaluation strongly supports the diagnosis of a malignancy, particularly one for which a frozen section analysis will be difficult to do, an excisional biopsy should be considered. The advantages include single surgical procedure; however, a significant disadvantage is the need for extensive tissue sacrifice if re-excision is necessary (malignant tumor) to obtain appropriate margins (i.e., unplanned excision) (
Fig. 13-6). An added advantage of an excisional biopsy is that the pathologist is able to examine the entire lesion, thereby improving the accuracy of the pathologic examination. An incisional biopsy exposes uncontaminated tissues to the tumor, and if the tumor proves to be a malignancy, the definitive resection is more complicated. If the lesion can be treated with curettage or a marginal excision, the incisional biopsy leads to the least functional loss. The final decision is made for each patient on the basis of not only the tumor’s characteristics but also the patient’s preference. Some patients want to take the fewest chances, and are willing to accept the possibility of slight overtreatment, whereas others choose to take one step at a time. It is the surgeon’s responsibility to explain the situation to the patient so that an informed decision can be made.
A final note of caution is offered with regard to the biopsy: osteomyelitis is more common than bone tumors, especially in children, and osteomyelitis often mimics neoplasia. The reverse is also true; therefore, when performing a biopsy, even
when the diagnosis seems obvious, culture every biopsy and biopsy every culture.