Chapter 5 Biomarkers of Disease Activity
BIOMARKERS AND SURROGATE ENDPOINTS
The terms biomarker and surrogate endpoint describe different entities. However, they are commonly used interchangeably, which has led to considerable confusion in the literature. In an attempt to prevent such confusion and standardize the nomenclature, the National Institutes of Health convened an expert panel in 1991. Their recommendations are summarized in Table 5.1.1 Biomarker can be defined as a physical sign or cellular, biochemical, molecular, or genetic alteration by which a normal or abnormal biologic process can be recognized and/or monitored, and that may have diagnostic or prognostic utility. Biomarkers must measure an underlying biologic process reliably and reproducibly. Surrogate endpoint is a measurement that is intended to serve as a substitute for a clinically meaningful outcome and is expected to predict the effect of a therapeutic intervention. Both biomarkers and surrogate endpoints have to be validated to prove that they are measuring intended outcomes reliably. It is important to recognize that the requirements of surrogate markers are much more stringent and that only a small minority of biomarkers will fulfill the criteria of a surrogate endpoint. In order for a biomarker to be validated as a surrogate endpoint, it must be shown that the presence of or a change in the measurement predicts an important clinical endpoint.
TABLE 5.1 VALIDATION OF BIOMARKERS AND SURROGATE ENDPOINTS
VALIDATION OF BIOMARKERS
Validation of biomarkers is a complex process.2,3 The criteria for validation should be defined by the nature of the question that the biomarker is intended to address, the degree of certainty required for the answer, and the assumptions between the biomarker and clinical endpoints. An ideal biomarker should measure a clinically relevant process, and be sensitive and specific for the measurement that it is intended for.
Any biomarker should be validated for sensitivity, specificity, details of bioanalytical assessment, and the probability of false positives and false negatives. Sensitivity is the ability of a biomarker to reflect a meaningful change in important clinical and/or biological endpoints and describes the level of correlation between the magnitude of change in the biomarker and clinical/biological endpoint. However, even a strong correlation does not prove a cause–effect relationship. Specificity defines the extent to which a biomarker explains the changes in a clinical/biological endpoint. The bioanalytical assessment of the laboratory test or measurement should include assessment of precision, reproducibility, range of use, variability, and practicality. False positivity is the situation in which a desired change in a biomarker is not reflected by a positive change in a clinical/biological endpoint, or even worse, is associated with a negative change. False negativity is the opposite, when a biomarker does not change despite a change in the clinical/biological outcome (Table 5.1).
Qualification of Surrogate Endpoints
An ideal surrogate endpoint can be thought of as a validated biomarker that can be definitively substituted for a clinically meaningful endpoint in an efficacy trial or clinical practice. To meet the most rigorous standards, the surrogate endpoint must correlate with the true clinical outcome and must fully capture the net clinical effect of treatment.4 This may be impossible to achieve for most biomarkers, but it is clear that extensive clinical evidence is needed, which is collected in a rigorous scientific process and analyzed by careful statistical assessment. There is no consensus on validation of surrogate endpoints, and some experts favor the term qualification to describe this process,1,5 which has to include the following elements. Biologic plausibility should provide a mechanistic basis for using a surrogate endpoint, and epidemiologic or observational studies of the natural history of the disease should establish the statistical relationship between the biomarker and the clinical endpoint. Adequate, well-controlled and appropriately powered clinical trials should provide an estimate of the expected benefit; ideally, an appropriate dose– or exposure–response relationship would provide additional support for surrogate status. The analysis should include a consideration of whether potential adverse reactions are predicted by the surrogate endpoint. It is essential that the development and validation of biomarkers and surrogate markers be built into the drug development process, starting from the preclinical phase. It may be helpful to conduct a meta-analysis of multiple clinical trials to determine the consistency of effects following interventions with various drug classes and within different stages of disease.2
Current State of Biomarkers in SLE
A large number of studies described potential biomarkers in lupus, but none fulfills the criteria of a true validated biomarker. There are many reasons that account for the conflicting results of various studies. Most were not designed as biomarker validation studies; therefore, their study design may not be appropriate for this purpose. A cross-sectional study may show an association between a biomarker and disease activity in a group of patients, but longitudinal studies are necessary to evaluate whether the same marker can be used to monitor disease activity in individual patients. The patient population may differ among studies in several ways such as ethnicity, treatment, organ manifestation, and stage of disease (early vs. late). The choice of controls (healthy vs. other rheumatic diseases vs. subpopulations of lupus patients) is also critical, and varies substantially among studies. Most frequently, widely accepted disease activity indices, such as SLEDAI, SLAM, ECLAM, and BILAG, are used as outcome measures. Although all are valid tools, they do not capture exactly the same aspects of lupus.6 Therefore, in some studies selected biomarkers correlated with one but not another activity index. In some cases, organ-specific outcome measures may be more appropriate, but the lack of widely accepted clinical endpoints makes standardization difficult. A large number of studies lack the statistical rigor that is essential to draw valid conclusions. Last, but not least, the bioassays used to measure biomarkers are frequently not standardized, leading to conflicting results in different laboratories.
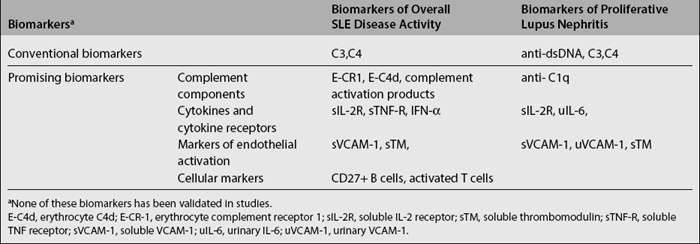
BIOMARKERS IN SLE
Lupus is a chronic disease with various mechanisms dominating at various stages. Genetic predisposition and environmental factors are the most important for predicting or quantifying the risk of lupus. At the onset of clinical symptoms, classic biomarkers of autoreactivity such as autoantibodies are used to help to establish or confirm the diagnosis of SLE. During the acute phases of lupus, biomarkers of disease activity could be used to optimize risk–benefit assessment in individual patients. Eventually, the sustained autoimmune and inflammatory process leads to the next phase characterized by organ damage, when there is a much greater reliance on physiologic measures of organ function, such as renal function measurements.
Due to the complexity of the underlying pathogenesis, it is likely that any specific biomarker will perform best at certain stages of the disease and novel biomarkers have to be defined specifically as to what they are intended to reflect (prognosis, future organ involvement, severity, disease activity, risk of flare-up, etc.), and at what stage of the disease (Table 5.2).
TABLE 5.2 POTENTIAL USES OF BIOMARKERS IN SLE
| Application | Potential Biomarkers |
|---|---|
| Predict SLE and/or organ involvement | |
| Monitor disease activity | |
| Predict response to therapya | |
| Predict flarea | Changes in selected markers of diseases activity |
| Predict damagea | |
| Describe damage |
a Therapeutic decision based on predictive biomarkers should be made only after a strong correlation between the biomarker and the clinical outcome has been established; that is, the biomarker is qualified as a surrogate endpoint.
A detailed review of the literature on novel biomarkers in SLE was published recently.7,8 Other chapters in this book describe the genetics of lupus and the current use of various laboratory tests in diagnosing SLE and their relationship to specific clinical manifestations. Here we will provide a summary of both the conventional and the most promising biomarkers of disease activity.
Recent studies have demonstrated that autoantibodies can be found in the majority of lupus patients years before the diagnosis or the onset of symptoms.9,10 Moreover, the appearance of antibodies seemed to follow a temporal sequence. In 88 of the 130 patients with SLE, at least one autoantibody tested was present before the diagnosis (up to 9.4 years earlier; mean, 3.3 years). High titer antinuclear antibodies were present in 78%; anti–double-stranded DNA and anti-Ro antibodies in about 50%; anti-La, anti-Sm, and antinuclear ribonucleoprotein in about 30%; and antiphospholipid antibodies in 18% of patients. Antinuclear, antiphospholipid, anti-Ro, and anti-La antibodies were present earlier than anti-Sm and antinuclear ribonucleoprotein antibodies (a mean of 3.4 years before diagnosis vs. 1.2 years). Anti–double-stranded DNA antibodies, with a mean onset 2.2 years before the diagnosis, were found later than antinuclear antibodies (p=0.06) and earlier than antinuclear ribonucleoprotein antibodies. The finding that the appearance of autoantibodies in patients with SLE tends to follow a predictable course, with a progressive accumulation of specific autoantibodies before the onset of SLE raises the possibility of using autoantibody profiles as predictors of lupus. These observations are clearly exciting; however, their clinical applicability needs to be determined.
Although they account only for a very small proportion of patients, deficiencies in early complement components, such as C4, C2, and C1q, are associated with a significantly increased susceptibility to lupus. Moreover, activation of the complement system plays a central role in the pathogenesis of lupus. Therefore, it is conceivable that subtle changes in the complement system could be used for early diagnosis of SLE. A recent study showed that combined detection of high levels of erythrocyte-bound C4d (E-C4d) and low levels of erythrocyte-complement receptor 1 (E-CR1) had a sensitivity of 81% and specificity of 91% in distinguishing SLE patients from normal controls and acceptable sensitivity (72%) and specificity (79%) in distinguishing SLE and other autoimmune diseases. The overall negative predictive value of the combination of the two tests was 92%.11–13 Platelet C4d was found in only 18% of lupus patients, but it had 100% specificity against normal subjects and 98% against patients with other autoimmune diseases.14 Both of these findings have to be confirmed in larger studies, and their applicability to early diagnosis needs to be formally tested.
Markers of Disease Activity
Classical Markers of Lupus Activity
Despite a large body of literature about the associations of various autoantibodies with clinical manifestations and/or disease activity, there is remarkably little consensus on the value of these examinations in specific situations in individual patients; even the most widely used tests, such as anti-dsDNA antibodies and complement levels, are controversial. The opinions about the utility of anti-dsDNA ranges from proponents in favor of preemptive treatment in response to increases in anti-dsDNA15,16 to believers that such changes have no value in predicting flare-ups.17,18 Several recent publications and reviews addressed this issue.16–22 It is clear that methodologic differences, such as the frequency of testing, the tools used to assess activity, the definition of flares and the statistical methods used all contributed to the conflicting results. Furthermore, a 1-year longitudinal study of 53 patients found a decrease, and not an increase, in anti-dsDNA levels at the time of flares,23 presumably due to deposition of anti-dsDNA in tissues. Interestingly, flares measured by some but not all disease activity measures were preceded by an increase in anti-dsDNA levels.
Complement has an important role in the pathogenesis of SLE. Traditional measures of complement activity, such as CH50, C3, and C4, have low sensitivity and specificity because plasma levels reflect the result of the dynamic state of complement synthesis and consumption, both of which are increased during inflammation. Activation of the complement system is characterized by the generation of activated breakdown products of precursor molecules. Complement activation products may be more specific for complement activation and there is a good rationale to use them as markers of disease activity. However, the available studies show conflicting results with markers of the classic, alternative, or common pathways showing correlation with activity in some but not in other studies. Some of this may result from methodologic differences, such as the use of plasma versus serum and differences in the definition of disease activity. The instability and high turnover of the complement products have also limited their use as potential biomarkers. One direction for investigations in this field was an attempt to measure erythrocyte bound isoforms of complement products and complement receptor on the surface of erythrocytes and reticulocytes. These complement-split products are acquired on the surface of the red blood cells during activation of the classical pathway, and the accumulated change in their levels is thought to reflect the state of complement activation for as long as the life span of a normal erythrocyte.12 It has also been shown that patients with SLE have reduced clearance of immune complexes associated with decreased levels of complement receptor 1 (CR1) on erythrocytes.24 In a recent study, expression of erythrocyte bound C4d (E-C4d) and CR1 (E-CR1) was determined by flow cytometry in 100 patients with SLE, 133 patients with other diseases, and 84 normal controls.13 Patients with SLE had significantly higher levels of E-C4d and lower levels of E-CR1 than healthy controls or patients with other diseases. The two tests combined together could distinguish patients with SLE from healthy controls with 81% sensitivity and 91% specificity, and SLE from other diseases with 72% sensitivity and 79% specificity. Another two color flow–cytometric analysis of C4d and CR1 on the surface of reticulocytes performed by the same group of investigators in 156 SLE patients, 140 patients with other diseases, and 159 healthy controls demonstrated significantly higher levels of C4d in SLE when compared with the two other groups; lupus patients with reticulocyte C4d levels in the highest quartile had higher SELENA-SLEDAI and SLAM scores than patients whose reticulocyte C4d levels were in the lowest quartile.11 Further work and large-scale trials are needed in this area to help further define appropriate complement-split products for assessing lupus disease activity, and to determine whether any of these can be used as a reliable biomarker.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


