Chapter 13 Biology of Dendritic Cells
INTRODUCTION
The primary role of the immune system is to protect the body against infections. Innate (non Ag-specific) and adaptive (Ag-specific) immunity cells act in concert with effector proteins such as cytokines, antimicrobial peptides, complement, and antibodies to eradicate pathogens.1–3 Lymphocytes (T cells, B cells, NK and NK T cells) and their products are under the control of dendritic cells (DCs).4–7 DCs are composed of multiple subsets with distinct functions. These cells populate peripheral tissues where they capture antigens (Fig. 13.1). Antigen-loaded DCs migrate from the tissues through the afferent lymphatics into the draining lymph nodes. There, they present processed protein and lipid Ags to T cells via both classical (MHC class I and class II) and nonclassical (CD1 family) antigen-presenting molecules.6 Immature (nonactivated) DCs present self-antigens to T cells8–10 and, in the absence of appropriate co-stimulation, induce tolerance. Mature, antigen-loaded DCs induce antigen-specific immunity11 characterized by T-cell proliferation and differentiation into helper and effector cells with unique function and cytokine profiles. DCs can also induce immune tolerance through T-cell deletion and through activation of regulatory T cells. How this complex balance is maintained in health and broken in disease, and how it is regulated through distinct DC subsets and their functional plasticity, is now starting to be understood. We will focus on recent progresses in our knowledge of the physiology of DCs, on the identification of distinct DC subsets that induce distinct types of immune response6,12,13 and how this information might impact our understanding of autoimmune diseases like SLE.
BIOLOGY OF DENDRITIC CELLS
DCs Capture and Present Antigens
Immature DCs are remarkably efficient at Ag capture while mature DCs are remarkably efficient at antigen presentation. DCs use several pathways to capture antigen, including (1) macropinocytosis; (2) receptor-mediated endocytosis via C-type lectins (e.g., mannose receptor, DEC-205, DC-SIGN)14–21 or Fcγ receptors type I (CD64) and type II (CD32) (uptake of immune complexes or opsonized particles)22; (3) phagocytosis of apoptotic and necrotic cells,8,9,23 viruses, and bacteria including mycobacteria,24,25 as well as intracellular parasites such as Leishmania major; and (4) internalization of heat shock proteins, hsp70 or gp96-peptide complexes, through multiple receptors including LOX-126 and TLR2/4.27 Captured antigens are processed in distinct intracellular compartments and loaded onto DCs antigen presenting molecules (reviewed in Trombetta and Mellman28). Protein antigens are presented by classical MHC class-I and class-II molecules, while lipid antigens are presented through nonclassical CD1 antigen-presenting molecules.6
Presentation via MHC Class II and Class I
Captured antigens are presented by MHC class-II molecules,29 which upon DC maturation are transported from lysosomal compartments to the cell membrane.30,31 In fact, this translocation of peptide-MHC (pMHC) class-II complexes from intracellular compartments to cell membrane represents a hallmark of DC maturation. pMHC complexes are very stable on the cell membrane of mature DCs, thereby facilitating TCR recognition. Furthermore, as opposed to macrophages that favor antigen degradation, DCs display lower levels of lysosomal proteases, thereby permitting low rate of antigen degradation.32 This in turn allows antigen retention in lymphoid organs in vivo for extended periods that might favor antigen presentation.32 Thus, prolonged availability of antigen for generation of pMHC complexes and prolonged presentation of such complexes on the cell surface might explain the unique efficiency of DCs in triggering naive T-cell differentiation.
MHC class-II molecules are under the control of a transcriptional coactivator, MHC class-II transactivator (CIITA).33 The expression of CIITA is regulated by three independent promoters, the activity of which quantitatively determines MHC class-II expression.34 Distinct subsets of antigen-presenting cells utilize different promoters, that is, plasmacytoid DCs (pDCs) and B cells rely on promoter pIII while myeloid DCs (mDCs) and macrophages use pI.35 These differences may have fundamental impact on the antigen presentation on MHC class II by these cell types and on ensuing immune responses.
MHC class-I molecules represent another antigen-presentation pathway exploited by DCs.28 This involves the classical presentation of endogenous peptides, originating from cellular and viral proteins, as well as the presentation of exogenous antigens via cross-priming/presentation. In fact, cross-priming/presentation might be the main pathway through which immunity to tumors and microbes that do not infect DCs directly is generated.8,36,37 The cellular mechanisms that DCs use to present exogenous antigens on MHC class I remain to be defined. Cross-priming might actually depend on the transfer of proteasome substrates rather than peptides.38 Furthermore, the loading compartment remains as yet an unresolved issue, that is, whether it is the endoplasmic reticulum (ER) or a mixed phagosome-ER compartment.39
Presentation via CD1 Family of Nonclassical MHC Molecules
CD1 proteins present lipid antigens to effector T cells.40,41 In the human, but not in the mouse, this family consists of four members, CD1a-c (group 1 molecules) and CD1d (group 2 molecules), with distinct expression patterns.40,41 Different CD1 molecules display distinct intracellular trafficking pathways that likely result in antigen delivery into distinct compartments.40,41
CD1d is involved in antigen presentation to natural killer (NK) T cells, a unique subset of T cells expressing a limited TCR repertoire mostly composed of Vα24Vβ11.42 These innate-like T cells contribute to immune responses to infections and malignancies. Recent studies have identified lysosomal glycosphingolipid, isoglobotrihexosylceramide (iGb3),43 as endogenous antigens, and bacterial glycosylceramides44 as exogenous antigens for presentation by CD1d to NKT cells. Interestingly, CD1d ligation on monocytes triggers translocation of NFκB and IL-12 secretion,45 thus providing a possible mechanism through which NKT cells modulate antigen presenting cells and immune responses.46
DCs Migrate and Orchestrate Migration of Other Cells
Blood immature/nonactivated DCs are attracted to tissues through MIP3-a (via CCR6) or MCP chemokines (via CCR2), as demonstrated for Langerhans cells (LCs) in vitro47 and in vivo,48 respectively. Upon pathogen entry, epithelial cells secrete ligands that provide signals for enhanced DC influx. Indeed, DCs are the first cells to arrive at the site of pathogen entry, even preceding neutrophils.49–51 Little is known about how DCs enter and traffic through the lymphatic vessels. The chemokine receptor CC-chemokine receptor 7 (CCR7) appears to be fundamental in this process.52 Thus, distinct maturation/activation signals—for example, prostaglandin PGE253,54 that induce the preferential expression of CCR7 by DCs—might increase the capacity of DCs to respond to appropriate ligands such as CCL19 and CCL21 expressed in lymphatic vessels and secondary lymphoid organs.55
Distinct DC subsets might enter secondary lymphoid organs through distinct routes. Indeed, upon bacterial triggering, mDC precursors migrate to peripheral tissues and subsequently to draining lymph nodes, while pDC precursors directly enter the lymph nodes in a CXCL9 and E-selectin dependent manner.56 Differential migration of cutaneous DC subsets, that is, LCs and dermal DCs, has been demonstrated recently with the use of vital imaging.57 Thus, after skin immunization, dermal DCs arrived in lymph nodes first and colonized areas distinct from those colonized by the slower-migrating LCs.57 These results might shed light on the way an immune response develops upon cutaneous immunization, a classical route of vaccination. DCs not only migrate, but they orchestrate the migration of immune effectors through chemokines,58–64 and regulate their maturation and function through cell-cell contact and/or soluble factors.4–7,65
DC Maturation
DC migration is intimately linked with maturation, and has an important impact on T-cell immunity.
Maturation Signals
DCs can receive maturation signals from (1) T cells through CD40 ligand,66 as well as from NK, NKT cells, and γ/δ T cells (reviewed in Munz et al.46); (2) proinflammatory molecules including IL-1β, TNF, IL-6, and PGE2,67 or a combination of IL-1β and TNF with type-I (IFN-α/β) and -II (IFN-γ) interferons68; and (3) DC surface molecules involved in pathogen recognition, including Toll receptors (TLRs) and C-type lectins (reviewed in Klechevsky et al.69). It is likely that in vivo DCs will be exposed to a combination of these signals, which then influence the net result of T-cell activation. Interestingly, TLRs are differentially expressed by distinct DC subsets. For example, TLR9 (a receptor for demethylated DNA) is expressed only by pDCs, while mDCs preferentially express TLR 2 and 4 (receptors for bacterial products such as peptidoglycan and lipopolysaccharide, respectively).70 Similarly, distinct DC subsets express unique lectins,20 which display immunostimulatory (ITAM) or inhibitory (ITIM) motifs. Such differential expression may confer distinct maturation signals and yield a distinct type of immune responses.71 TLR-mediated signaling in other cells (i.e., stromal cells) will trigger expression of specific chemokines/cytokines and adhesion molecules, which in turn will indirectly modulate DC maturation.72
Maturation Phenotype
As DCs mature, a series of coordinated events take place such as (1) loss of endocytic/phagocytic receptors; (2) up-regulation of co-stimulatory molecules CD40, CD80, CD86, and several members of the TNF/TNF receptor family including CD70 (ligand for CD27), 4-1BB-L, and OX40-L, all of which can have co-stimulatory effects on T cells73; (3) changes in morphology that include the loss of adhesive structures, cytoskeleton disorganization, and acquisition of high cellular motility74; (4) shift in lysosomal compartments with down-regulation of CD68 and up-regulation of DC-LAMP75; (5) change in class-II MHC compartments; and (6) secretion of cytokines including IL-12 and IL-23, which are important for type-1 polarization of T-cell immunity.
This basic process of DC maturation can be modulated by pathogens via interaction with TLRs expressed on DCs. For example, TLR ligands together with a T-cell-like signal delivered through CD40, may enhance DC function.76 Indeed, TLR-mediated signals are involved in the control of CD4+ T-cell activation,77 and may play a role in autoimmunity. For example, DCs loaded with a heart-specific self-peptide induce CD4+ T-cell-mediated myocarditis in nontransgenic mice if activated through both CD40 and TLRs.78
Functional DC maturation can also be modulated by C-type lectins. Dectin-1, a yeast binding C-type lectin, synergizes with TLR2 to induce TNF-α and IL-12 production.79 Yet, Dectin-1 can also promote synthesis of IL-2 and IL-10 through recruitment of Syk kinase. Accordingly, syk-/- DCs do not make IL-10 or IL-2 upon yeast stimulation, but produce IL-12, which indicates that the Dectin-1/Syk and Dectin-1/TLR2 pathways can operate independently.79 These results demonstrate that pathogens use several surface molecules to modulate DC function.
Dendritic Cells Determine Type of T-Cell Response
DC Maturation and Outcome of Interaction with T Cells
It is currently thought that immature DCs are tolerogenic, while mature DCs are immunostimulatory.80 This has been formally demonstrated in vivo with the use of fusion proteins targeted to immature DCs that lead to the induction of antigen-specific tolerance.81 By contrast, concomitant activation of the DCs with a CD40-specific antibody results in a potent immune response because DCs are induced to express a large number of co-stimulatory molecules.82 Thus, immature DCs in the steady state are thought to maintain peripheral tolerance through their ability to present tissue antigens without appropriate co-stimulation. However, mature LPS-activated DCs efficiently expand CD25+CD4+ regulatory T cells.83 Additionally, LCs reaching the lymph nodes under steady-state or inflammatory conditions have been found to express similar levels of MHC class II, CD40, and CD86.57 Thus, the outcome of tolerance induction or priming might not only be related to the maturation stage, but to specific maturation signals, the threshold of activation,84 and/or the activation of a unique set of inhibitory molecules on DCs.
DC Maturation and Regulatory/Suppressor T Cells
Two broad subsets of CD4+ T cells with regulatory function have been characterized,85–87 both of which can be activated/expanded by DCs at distinct maturation stages.
Naturally Occurring CD4+CD25+ T Cells
These T cells are produced in the thymus and mediate their suppressive effects in a cell-contact-dependent, antigen-independent manner, without the requirement of IL-10 or TGF-β.88–91 These cells are naturally “anergic” and require stimulation via their TCR for optimal suppressive function. Mature DCs allow their expansion, which is partially dependent on the production of IL-2 by the T cells and B7 co-stimulation by the DCs.83
Induced T-Regulatory Cells
T-regulatory (TR) cells derive from CD4+25− T cells and mediate their effects through the production of suppressive cytokines such as IL-10 and TGF-β.92–94 Two types have been described: TR1 cells produce large amounts of IL-10 and low to moderate levels of TGF-β,92 and Th3 cells produce preferentially TGF-β95 and provide help for IgA production.96 Immature DCs induce the differentiation of naive T cells into TR cells.92,97,98 Injection of immature DCs pulsed with influenza-derived peptide has been shown in two healthy adults to lead to antigen-specific silencing of effector T-cell function.99 Murine pulmonary DCs induce the development of TR in an ICOS-ICOS-L-dependent fashion that leads to the production of IL-10 by DCs.100 Furthermore, a population of “semimature” CD45RBhigh CD11clow murine DCs located within the spleen and lymph nodes has been described. These cells secrete IL-10 after activation with LPS or CpG oligonucleotides, but do not up-regulate MHC class II or co-stimulatory molecules under the same conditions. Most importantly, they are highly potent at inducing tolerance that is mediated through the differentiation of TR cells in vivo.98,100 The complexity of the lineage and/or the subpopulations of DCs that may be responsible for tolerance induction is further illustrated by the description of unconventional DCs101 that display phenotypic and functional properties of both natural killer (NK) and dendritic cells (DCs). These cells appear able to induce protection against virally induced type-1 diabetes in a mouse model.
DC Subsets and Type of Induced T-Cell Immunity
Finally, distinct DC subsets differentially modulate T-cell immunity. In mice, splenic CD8α+ DCs prime naive CD4+ T cells to make Th1 cytokines in a process involving IL-12, whereas splenic CD8α− DCs prime naive CD4+ T cells to make Th2 cytokines.102,103 Furthermore, different signals can induce different T-cell polarization by the same DCs, as shown by the induction of IL-12 production and Th1-cell polarization when DCs are activated with Escherichia coli LPS, but no IL-12 production and Th2-cell polarization when DCs are exposed to LPS from Porphyromonas gingivalis.104 In humans, CD40-ligand (CD40-L)– activated, monocyte-derived DCs prime Th1 responses through an IL-12-dependent mechanism, whereas pDCs activated with IL-3 and CD40-L have been shown to secrete negligible amounts of IL-12 and prime Th2 responses.105 Furthermore, IL-3- and CD40-L–activated pDCs induce CD8+T cells with regulatory/suppressor function.106,107 Thus, both the type of DC subset and the activation signals to which DCs are exposed are important for T-cell polarization.
Dendritic Cells Are Composed of Subsets
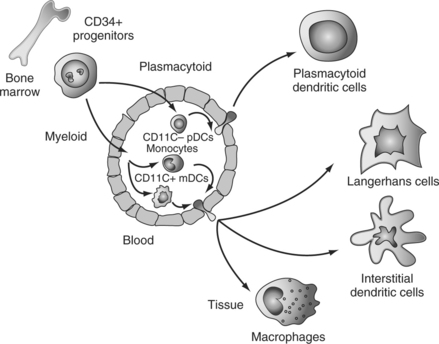
Classically, two main DC differentiation pathways are recognized.6,7 A myeloid pathway generates Langerhans cells (LCs), which are found in stratified epithelia such as the skin, and interstitial (int)DCs, which are found in all other tissues.108 Another pathway generates plasmacytoid DCs (pDCs),109 which secrete large amounts of IFN-α/β after viral infection.110–112
DC Progenitors and Precursors
DC progenitors reside within CD34+ hematopoietic progenitor cells (HPCs).113 Both lymphoid and common myeloid progenitors yield, at the clonal level, mDCs as well as cells with pDC phenotype and capacity to secrete large amounts of IFN-α.114 Interestingly, the progenitors of pDCs and mDCs can be found within FLT3+ HPCs.114,115 This is consistent with the well-established role of FLT3 ligand (FLT3-L) in DC differentiation/mobilization in vivo in both humans and mice.117–120 Accordingly, FLT3-L is essential in the generation of pDCs and myeloid DCs (mDCs),122–124 and FLT3-L deficient mice show a considerable decrease in numbers of DCs in both peripheral and lymphoid tissues.117 Thus, FLT3-L appears as a major factor governing DC homeostasis in the steady state. Given the role of GM-CSF in DC generation,113,125,126 activation and survival,127 it is possible that GM-CSF is actually a major factor governing DC homeostasis upon infection.
Until recently monocytes, lineage− mDC precursors and pDCs have been considered as major circulating DC precursor populations (Fig. 13.1). However, recent studies demonstrated that γ/δ T cells can acquire DC phenotype and function.128 Furthermore, proinflammatory cytokines can endow human NK cells with the ability to acquire antigen and stimulate T cells.129 These observations suggest remarkable plasticity in the antigen-presenting cell system. Thus, monocytes may yield all myeloid DCs while pDCs, γ/δ T cells, and NK cells yield another set of cells with DC properties.
Myeloid DC Diversity
This concept of plasticity/flexibility of the DC system is even further exemplified by monocytes and their response to environmental signals. Thus, monocytes can differentiate into either macrophages, which act as scavengers, or DCs that induce specific immune responses.130,131 Different cytokines skew the in vitro differentiation of monocytes into DCs with different phenotypes and function. Thus, when activated (e.g., by GM-CSF) monocytes encounter IL-4, they will yield IL4-DCs.132–134 By contrast, after encountering IFN-α/β, TSLP, TNF, or IL-15, activated monocytes will differentiate into IFN-DCs,135–138 TSLP-DCs,139,140 TNF-DCs,141 or IL15-DCs,142 respectively. Thus, rather than the classical distinction of LCs and intDCs, we should consider myeloid DCs as a gradient of immunostimulatory DCs. Thus, mDCs are polarized by other cells and their products including IFN-α from pDCs, IFN-γ from γ/δ T cells and NK cells, IL-4 and TNF from mast cells, IL-15 and TSLP from stromal cells, and IL-10 from lymphocytes. These distinct DCs will induce distinct types of T-cell immunity. The challenge for years to come will be to link these distinct DC phenotypes in vitro with a specific type of immune response and immune pathology in vivo as exemplified by TNF and IFN-α143,144 or by TSLP in allergic inflammation.139
Distinct DC Subsets Are Endowed with Distinct Functional Properties
Each DC subset has common as well as unique biological functions determined by a unique combination of cell-surface molecules and cytokines. Thus, in vitro experiments showed that LCs and interstitial DCs generated in cultures of CD34+ hematopoietic progenitors differ in their capacity to activate lymphocytes: interstitial DCs induce the differentiation of naive B cells into immunoglobulin-secreting plasma cells,108,145 whereas LCs seem to be particularly efficient activators of cytotoxic CD8+ T cells. They also differ in the cytokines that they secrete (e.g., only interstitial DCs produce IL-10) and their enzymatic activity,108,145 which might be fundamental for the selection of peptides that will be presented to T cells. Indeed, different enzymes are likely to degrade a given antigen into different peptide repertoires, as recently shown for HIV nef protein.146 This will lead to different sets of pMHC complexes being presented and to distinct antigen-specific T-cell repertoires.
DC subsets express unique lectins,20 which at least partially account for the biological differences. Thus, LCs express langerin, critical to the formation of Birbeck granules.147,148 The role of these structures is not yet understood. The intDCs express DC-SIGN, which is involved in the interactions with T cells and DC migration, but is also used by pathogens, such as HIV, to hijack the immune system.149–151 pDCs express yet another lectin BDCA2, which is absent on mDCs.152,153 TLRs are also differentially expressed, as only pDCs express the nucleic acid-recognizing TLR-7/8 and TLR-9. Such differential expression may permit specific in vivo targeting of DC subsets for induction of a desired type of immune responses, as recently demonstrated in mice by targeting DEC-205.81,82
Importantly, we begin to understand the molecular pathways underlying differential responses of pDCs and mDCs to pathogens and/or pathogen derived factors. This can be best illustrated by studies on mechanisms regulating type-I interferon secretion. Thus, pDCs are recognized as a main source of type-I interferon produced in response to viral110 or CpG154,155 triggering. Recent studies demonstrated that IRF-7 is critical for IFNα/β secretion in response to both stimuli. However, IRF-7 activation in response to virus is MyD88 independent, while response to CpG is dependent on both IRF-7 and MyD88.157 It turns out that pDCs but not mDCs can direct CpG to endosomal compartments, thereby allowing MyD88/IRF-7 activation and IFNα/β secretion.157
Subsets of pDCs
pDCs display distinct functions at two distinct stages of differentiation: (1) precursor pDC secrete large amounts of IFN-a after viral infection,110–112 and (2) mature pDC activate and modulate T-cell responses.158 Indeed, depending on the type of activation pDCs give rise to unique T-cell responses: they induce T cells secreting IFN-γ and IL-10 upon viral triggering and type-2 T cells upon activation with IL-3 and CD40 ligand.158 Recently the existence of pDCs subsets has been demonstrated. Thus, in the mouse, expression of lymphoid-related genes (RAG1 and Ig rearrangement products158) or proteins (CD4159) distinguishes between two subsets of pDCs. While the functional consequence of RAG1 and Ig rearrangement product expression remains to be determined, CD4neg pDCs appear mainly responsible for migration to lymphoid tissue and IFN-α secretion upon exposure to CpG.159
DCs Interact with Other Cells of Immune System
DCs also regulate naive145 and memory160 B cells, NK cells,161 and NKT cells.162
Interaction with B Cells
As discussed above, myeloid DCs can prime naive B cells. Several molecules have been shown to be involved in this process, including IL-12, IL-66 and, more recently, BAFF/Blys,163–167 a molecule up-regulated by IFN-α. IFN-α and IL-6 are also important in the differentiation of activated B cells into efficient Ig-secreting plasma cells upon exposure to virus-triggered pDCs.160 Strikingly, the plasma cells generated under these conditions express very high levels of CD38, similar to that of plasma cells isolated from lymphoid tissues. In contrast, plasma cells generated by culturing activated B cells with the T-cell-derived cytokines IL-2 and IL-10, although efficient Ig secretors, do not express high levels of CD38.168 This suggests that IFN-α may represent an important cytokine in the generation of plasma cells in tissues. Indeed, studies in the mouse also support that IFN-α is an excellent adjuvant for humoral immunity.169 However, myeloid DCs may also indirectly contribute to plasma cell differentiation.170–173 Finally, differential activation of CD4+T cells with B-cell helper function by distinct DC subsets might play an important role in the induction of protective humoral immunity.
Related posts:
 Assessment of Disease Activity in Systemic Lupus Erythematosus
Assessment of Disease Activity in Systemic Lupus Erythematosus
 The Environment in the Pathogenesis of Systemic Lupus Erythematosus
The Environment in the Pathogenesis of Systemic Lupus Erythematosus
 Antibodies and their Antigenic Targets in the Antiphospholipid Syndrome
Antibodies and their Antigenic Targets in the Antiphospholipid Syndrome
 Complement Deficiencies in Human Systemic Lupus Erythematosus (SLE) and SLE Nephritis: Epidemiology and Pathogenesis
Complement Deficiencies in Human Systemic Lupus Erythematosus (SLE) and SLE Nephritis: Epidemiology and Pathogenesis
 What Do Mouse Models Teach Us about Human Systemic Lupus Erythematosus?
What Do Mouse Models Teach Us about Human Systemic Lupus Erythematosus?
 Infection and Autoimmunity
Infection and Autoimmunity
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


