Chapter 11 B-cells in Human Systemic Lupus Erythematosus
Among the numerous immunologic abnormalities encountered in patients with SLE, the most striking is B-cell overactivity. B lymphocytes in SLE produce an array of autoantibodies (autoAb) against soluble and cellular constituents but most commonly and most characteristically against the macromolecular complexes of the cell nucleus, the antinuclear antibodies (ANA). Although the spectrum of autoAb specificity in SLE may seem unrestricted, only a handful of autoAB have been shown to contribute convincingly to disease-related tissue injury. The latter are best represented by the anti–blood-cell antibodies that activate complement and cause cytopenias and the cationic anti-dsDNA autoAb that are thought to contribute to the expression of nephritis.1 Apart from producing Ab, B-cells play a role in such things as the production of cytokines, antigen presentation, and immune regulation. Abnormalities of B-cell functions encountered in patients with SLE are discussed in this chapter.
THE ORIGIN OF AUTOREACTIVE B-CELLS
In contrast to previous beliefs, it is currently understood that autoreactive B-cells do exist in normal individuals. Moreover, although the naturally autoreactive B-cell pool was thought to be small, novel studies have presented evidence that this is an underestimation. Under normal conditions, or in disease states ranging from viral infections to malignancies, normal autoreactive B-cells produce a variety of natural autoAb that have a short life span and do not cause autoimmune disease or tissue damage. Natural autoAb usually belong to the IgM isotype, do not undergo isotype switching and affinity maturation, and may help in removing dying or dead cells. Still, under conditions of hypoxic tissue damage they may bind to newly revealed antigens, activate complement, and confer extensive tissue damage.2 In contrast, autoAbs in SLE undergo isotype switching and affinity maturation. Their presence is not helpful to the host because they have the capacity to cause tissue damage and their production is continuous and their presence is long lived, indicating that the relevant regulatory mechanisms are profoundly defective.
Autoreactive antibodies arise from autoreactive B-cells, but the mechanisms involved in the preservation of autoreactive B-cells are unclear. If we consider that detection of autoAb witnesses the presence of autoreactive B-cells, we now know that such B-cells exist in patients with SLE several years before the development of clinically evident disease. It was recently reported that 88% of previously healthy individuals that subsequently developed SLE have detectable autoAb in their sera as early as 9.4 years (mean: 3.3 years) before diagnosis, and in some perhaps even earlier. AutoAb were detected in the sera of 3.8% of healthy control individuals.3
The exhaustive analysis of different early B-cell subpopulations in a few patients with juvenile-onset SLE disclosed that autoreactive B-cells in SLE arise early in B-cell ontogeny and that the tolerance checkpoints imposed during B-cell development are violated. Significant numbers of antigen-inexperienced naïve B-cells (25 to 50%) are capable of producing autoAb in patients with SLE, whereas in healthy individuals this percentage does not exceed 20%.4
Despite the striking B-cell overactivity encountered in patients with SLE, challenging patients with standard immunizations or stimulating SLE peripheral B-cells with polyclonal activators in vitro can result paradoxically in substantially decreased amounts of specific Ab production compared to the responses of B-cells obtained from normal individuals5 (Fig. 11.1).
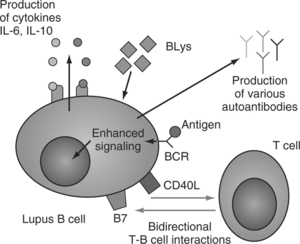
B-CELL SUBPOPULATIONS
Peripheral B-cells are phenotypically distinguished into the CD5+ B-1 cells (less than 20% of circulating B-cells) and the conventional B-cells (B-2 cells). B-1 cells may be involved in the pathophysiology of some human autoimmune diseases such as Sjögren’s syndrome and rheumatoid arthritis because they produce IgM rheumatoid factor. The potential pathogenic role of B-1 cells in human lupus is questionable principally because in patients with SLE both B-1 and B-2 cells contribute to the production of pathogenic autoAb such as anti-dsDNA.6
A detailed analysis of circulating B-cells in patients with SLE revealed major disturbances in the different peripheral B-cell compartments. A B-cell subpopulation representing immature B-cells (generated in the bone marrow but not having undergone final maturation steps in peripheral lymphoid organs) named type I transitional (T1) B-cells are reportedly increased in the circulation in patients with SLE.7 In patients with active (but not in those with inactive) SLE there was a marked reduction in the numbers of naive (CD19+CD27−) B-cells and an enhanced representation of the CD27highCD38+CD19dimsIglowCD20−CD138+ plasma cells in the periphery.8 In addition, increased numbers of CD27high plasma cells correlated with increased disease activity more accurately than a combination of clinical and serological parameters, suggesting that this may be of value in monitoring disease activity.9 Immunosuppressive therapy commonly used in SLE patients induces differential changes in the different B-cell subsets: it reduces significantly the numbers of CD27high plasma cells and of CD27− naive B-cells but does not affect the population of memory CD27+ B-cells.8 A similar reduction of both naïve and memory B-cells and a significant expansion of plasma-cell precursors is encountered in pediatric SLE patients. These B-cell subset alterations were independent of disease activity.10 Studying the development and fate of specific autoreactive B-cells (the natural autoAb VH4.34-producing B-cells) in patients with SLE and healthy individuals disclosed that in healthy donors the naive VH4.34 B-cells are numerous. However, following a sequence of selection steps in secondary lymphoid organs their progression into the final steps of differentiation is efficiently blocked. In contrast, naive VH4.34 B-cells in patients with SLE progress unopposed to the final steps of maturation into plasma cells. This suggests that tolerance checkpoints in patients with lupus are malfunctioning.11
THE ABNORMAL IMMUNOREGULATORY ENVIRONMENT
Isotype switching and affinity maturation (both features of lupus autoAb) indicate that the lupus autoAb response is a T-cell dependent (auto) antigen-driven immune process. But is the T-cell compartment responsible for B-cell hyperreactivity? If this is correct, B-cells should be under either decreased T-cell–mediated suppression or under excessive unopposed T-cell–derived help, or both. There is not much evidence supporting a decreased suppression, but there is evidence that increased T-cell–derived help is responsible for the increased production of antibody and autoAb in SLE. Increased nonspecific help to autoreactive B-cells can also lead to autoimmunity. A chronic graft-versus-host murine model supports this view12 and may provide an explanation for the increased occurrence of autoimmunity in patients undergoing bone marrow transplantations.13
INTRINSIC SLE B-CELL DEFECTS: FUNCTIONAL STUDIES
A number of studies have concluded that the B-cell in lupus may contribute in a T-cell–independent manner to the production of autoAb. Polyclonal B-cell activation is commonly encountered but is not the principal cause of the production of autoAb.14 According to the “two-signal” hypothesis, contact of B-cells with (self) antigen in the absence of T-cell–derived help tolerizes B-cells15 but in the presence of T-cell–derived help at the same time results in B-cell tolerance breakdown. Nevertheless, experimental data support the contention that this rule can be violated and that B-cell tolerance can be broken without the support of T-cell-mediated help. Such data have been produced in lupus-prone mice genetically manipulated to have no TCRαβ+ (and no CD4+) T-cells or even no T-cells at all. The B-cells of such autoimmunity-prone animals were still able to produce significant amounts of pathogenic IgG autoantibodies16 and to develop immune-complex–mediated nephritis,17,18 making the role of increased T-cell–mediated help or decreased suppression of relative importance.
Do overactive B-cells contribute to the expression of the disease directly, or through the production of autoAb? Lupus-prone MRL/lpr mice genetically manipulated to have B-cells incapable of secreting immunoglobulin still developed autoimmunity characterized by nephritis with cellular infiltrates, indicating that inherently autoimmune B-cells (without the production of autoAb and immune complexes) contribute directly to disease pathogenesis.19 The absolute dependence of the expression of SLE on either direct or indirect interactions of B-cells with either hyperactive or autoreactive T-cells is therefore uncertain under certain experimental conditions.
SLE B CELLS ARE EFFICIENT (AUTO) ANTIGEN-PRESENTING CELLS
Other investigators taking into account the particularly efficient antigen-presenting capacity of the B-cell have produced data indicating that the B-cell in lupus may represent the central pathogenic cell that triggers other immune cells toward hyperactivity. In certain experimental lupus models, the presence of B-cells even in the total absence of autoAb was the one and only determinant for the appearance of the autoimmune murine syndrome.19 In the absence of B-cells such autoimmune mice had neither activated T-cells nor lupus.20 It is thus possible that under certain circumstances lupus B-cells are not restricted to the production of autoAb only but mediate abnormal T-cell activation.21
In patients with SLE treated with anti-CD20 monoclonal antibody, the clinical effect was associated with decreased numbers of circulating T-cells expressing the early activation marker CD40L,22 suggesting that (as in mice) in humans lupus B-cells are involved in the generation of activated T-cells.
CYTOKINES CONTRIBUTE TO B-CELL ACTIVATION IN SLE
Human B-cells are capable of producing and secreting cytokines and lupus B-cells display an abnormally increased production of and response to IL-6 and IL-10. Both IL-6 and IL-10 are B-cell stimulatory cytokines, whereas IL-10 also inhibits type-1 cytokine responses. Patients with active SLE have elevated serum levels of IL-6,23 increased numbers of IL-6-secreting cells, and increased IL-6 mRNA content in their peripheral blood mononuclear cells. B-cells from patients with SLE express constitutively cell-surface IL-6 receptors. IL-6 stimulates SLE B-cells in vitro to produce anti-DNA Ab. SLE patients have elevated serum levels of IL-10, increased numbers of IL-10-secreting cells, and increased IL-10 mRNA content in their peripheral blood mononuclear cells. Recombinant human interleukin-10 promotes (and an anti–IL-10 mAb inhibits) the in vitro production of autoantibodies from B-cells obtained from patients with SLE.24 Increased production of IL-10 is also found in B-cells from relatives of patients with SLE. It has been reported that circulating lupus CD5+ B-cells (B-1 cells) expressing the surface marker CD40L spontaneously produce IL-10.25




