Chapter 16 Autoantibodies in Systemic Lupus Erythematosus
INTRODUCTION
Overview of Autoantibodies in SLE
One of the hallmarks of systemic lupus erythematosus (SLE) is the presence of autoantibodies, over a hundred of which have been reported in the serum of these patients.1 The vast majority are found rarely in SLE, or indeed in other autoimmune diseases. Many are likely to be bystanders and represent epiphenomena, but good evidence suggests that some cause clinical disease. Autoantibodies found in more than 25% of patients with SLE include anti-ssDNA, anti-dsDNA, anti-Ro, anti-poly ADP ribose, and anti-histone/nucleosome antibodies and antiphospholipid antibodies.
Antibodies to dsDNA are found in 50 to 70% of patients,2 but are very rarely found in healthy individuals and in relatives of patients with SLE.3 Interestingly, in one study, anti-dsDNA antibodies were detected in the sera for up to 9.4 years prior to developing symptomatic SLE.4 Anticardiolipin antibodies were estimated to occur in about 44% of patients with SLE, and 34% were found to have a lupus anticoagulant in one large meta-analysis.5 These results imply that not all anti-dsDNA or antiphospholipid antibodies are pathogenic. This observation will be discussed in some detail later in this chapter.
Basic Immunoglobulin Structure
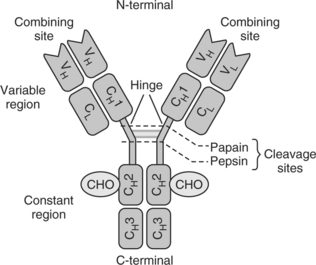
Before discussing the origin of autoantibodies in SLE, the structure of immunoglobulins must be considered. Antibodies, or immunoglobulins (Igs), play a pivotal role in the acquired immune response, and are produced by plasma cells in response to antigenic challenge. Igs consist of two heavy and two light chains, linked covalently by disulfide bonds (Fig. 16.1). The light chains may either be a kappa or lambda polypeptide and each of the two heavy chains contains four domains. It is the variable regions of the heavy and light chains at the N-terminal end that vary in amino acid sequence between antibody molecules and it is here that antigen binding occurs (Fab). The C-terminal end (Fc region) contains sites for complement fixation, and for example, binding to rheumatoid factors. Five classes of immunoglobulin are recognized on the basis of the Fc fragment of the heavy chain, and heavy-chain classes are further divided into subclasses. Idiotypic determinants are unique sequences of amino acids located in the variable region. It is here that antigen binding occurs and the site that determines antibody specificity.
Origin of Autoantibodies
The origin of autoantibodies in SLE is largely unknown but there are a number of theories. First, environmental autoantigens may trigger the induction of autoantibodies. Second, it has been shown in the MRL/pr mice models that anti-dsDNA production may be antigen driven.6 Similar findings and conclusions supporting antigen-driven autoantibody production have also been documented by other groups.7–9 Third, polyclonal B-cell activation has been suggested as a way of producing a diverse population of autoantibodies, supporting the idea of an important role played by B cells in SLE.10 Fourth, there are a number of ways in which apoptosis, or programmed cell death, may be involved in autoantibody production in this disease. Cellular debris produced as a result of apoptosis may act as the trigger for the production of autoantibodies. Since complement is required to aid in the clearance of apoptotic material, models of complement-deficient animals have confirmed that these animals are more prone to developing lupus-like nephritis.11 Other mechanisms of tissue injury include impairment of apoptosis, which in itself can be multifactoral and has been described elsewhere,12,13 and autoantibody-driven apoptosis.14,15 Many researchers are convinced that the failure of macrophages to efficiently remove apoptotic cells is critical.16 Apoptotic cell fragments containing nuclear “debris,” including DNA, Ro, La, Sm, and so on, may be “presented” to T cells by “professional” antigen-presenting cells or by B cells acting as antigen presenters. The T cells in turn trigger other B cells to produce anti-DNA, Ro, La, and so forth. In this way lupus is considered by some to be a consequence of inadequate “waste disposal.” Other mechanisms, including disruption of the idiotype network, may also play a role17,18.
STRUCTURAL CONSIDERATIONS
Anti-dsDNA Antibodies
Not all patients with these antibodies have overt clinical disease activity, suggesting that not all anti-dsDNA antibodies are pathogenic. This view has been confirmed in a number of animal studies whereby infusion of anti-dsDNA antibodies did not always result in disease.19–22 So what specific properties of anti-dsDNA antibodies make some but not others pathogenic? Furthermore, how and why do they develop and how do they cause tissue damage?
A considerable amount of work has been focused on attempting to answer these questions. For example, it has been established that IgG dsDNA antibodies tend to be the pathogenic isotype,23,24 which also tend to be positively charged and show high-affinity binding to dsDNA rather than ssDNA.2 The ability of some IgG anti-dsDNA antibodies to cross-react with a-actinin, which is found in the renal podocytes, may also be important.25
Role of Monoclonal Antibodies
One method of determining whether an autoantibody is pathogenic is to study the effect of human monoclonal antibodies in severe combined immunodeficiency (SCID) mice. Despite the difficulties in producing these monoclonal antibodies, various groups have made use of molecular biological techniques to synthesize a number of monoclonal anti-dsDNA and antiphospholipid (APL) antibodies, and their isotype and binding properties have been characterized. Describing these in any great detail is beyond the scope of this chapter, but they have been reviewed by Ravirajan and colleagues.26 The renal effects of a number of IgG human monoclonal anti-dsDNA antibodies that have been produced in SCID mice are shown in Table 16.1. Again it is clear that some antibodies are more pathogenic than others.
TABLE 16.1 IgG HUMAN MONOCLONAL ANTI-DNA ANTIBODIES Structure/function relationship EFFECTS IN SCID MICE
| Antibody | Proteinuria | Type of Deposition |
|---|---|---|
| B3 | ++→+++ | In vivo ANA |
| D5 | 0/+ | − |
| RH14 | ++→+++ | b.m capillary loop and mesangial matrix |
| DIL-6 | + | − |
| 32.B9 | + | − |
| 33.C9 | ++→+++ | Extracellular deposits in the capillary wall and mesangium |
| 35.21 | ++→+++ | In vivo ANA |
| C6.F7 | Trace | − |
Monoclonal anti-dsDNA antibodies from SLE patients27 and from animal lupus models28 have been sequenced, and analysis of their structure has shown that the high-affinity antibodies of the IgG isotype contain a large number of somatic mutations in their heavy-chain variable regions (VH) and light-chain variable regions (VL). These mutations are thought to be driven by antigens by virtue of their distribution within the complementarity-determining regions (CDRs). This region forms the antigen-binding site. Specific amino acids seem to be found more commonly in these CDRs as a result of somatic mutations, namely arginine, asparagine, and lysine. These amino acids are likely to be highly relevant by virtue of their ability to form electrostatic charges and hydrogen bonds with the negatively charged DNA phosphodiester backbone28,29 (see Fig. 16.1).
The human monoclonal anti-dsDNA antibody B3 has been extensively analyzed and shown to be pathogenic in SCID mice.30 Computer models31 have demonstrated the importance of its arginine residue at position 27a in CDR1 of the V γ gene 2a2. Substitution of this arginine by a serine both reduced the binding of the antibody to DNA and was associated with a significant reduction in proteinuria in the SCID model system.32
Many groups have looked at other autoantibodies and specific sequences in the heavy and light-chain variable regions and correlated this with DNA binding.33–35 Recent work has shown that particularly strong binding to dsDNA occurred when both light chains contained arginine residues (Arg 27a and Arg 92).35 They also showed changes in binding to DNA if arginine residues were positioned slightly differently. As reviewed elsewhere,36 most antibodies have more heavy-chain than light-chain contacts with DNA, especially in the heavy-chain third CDR. Perhaps there is a tendency for VH germline codons to mutate to arginine (or lysine), and a probability that B cells with arginine-containing CDR-VH domains will develop into anti-DNA antibody producers during clonal expansion.37
Kumar and colleagues38 investigated the human monoclonal autoantibodies B3, RH14 (anti-DNA), and UK4 (cardiolipin) in their IgG and cloned Fab formats, and looked to see whether they exhibited cross-reactivity and whether this might contribute to their pathogenic potential. The fine binding characteristics of these three molecules have been elucidated and published elsewhere, and will not be discussed further.26,39,40 Both B3 and RH14 antibodies have been shown to induce proteinuria in SCID mice, and RH14 shows specific renal binding and induces early features of lupus nephritis under electron microscopy.26,30 Kumar and colleagues38 showed that B3 and RH14 possess high cross-reactivity against a DNA polymerase (PolIV) of bacterial origin and that some patients with lupus have anti-PolIV activity in their sera. All three molecules bind to ssDNA and dsDNA, but RH14 is the predominant dsDNA binder and UK14 binds the most to cardiolipin. It is possible that changes in the physiologic milieu may modify the antigen-binding patterns of these antibodies, and thus modify their pathogenic potential, although this may be less true for anticardiolipin antibodies. Data from Kumar’s study also showed that the reactivity of RH14 for dsDNA involved high electrostatic charges, and phosphate interactions and B3 also significantly exhibited charge-independent cross-reactivity for cardiolipin.
Structural Consideration of Antiphospholipid Antibodies and Pathogenicity
Pathogenic anticardiolipin antibodies are also generally of the IgG isotype,41,42 and bind the cofactor β2-glycoprotein I (β2GPI). Analogous to the range of anti-dsDNA antibodies, it is interesting to note that patients with antiphospholipid antibodies do not always have the antiphospholipid antibody syndrome, characterized by arterial and venous thromboses, recurrent spontaneous miscarriage, and thrombocytopenia. Antiphospholipid antibodies have been found in other infectious and neoplastic diseases, as well as in normal healthy individuals.43 In general they are of the IgM isotype and do not bind β2GPI.
What seems to separate pathogenic from nonpathogenic antiphospholipid antibodies is the ability to bind neutral and negatively charged phospholipids in the presence of a co-factor, i.e., β2GPI.44 This co-factor is not the only one to adopt this “function,” and it has the ability to bind antiphospholipid antibodies in patients with the antiphospholipid syndrome even in the absence of phospholipids.45 Studies have shown similarities in the amino acid sequences of dsDNA antibodies and antiphospholipid antibodies, and arginine residues positioned at certain locations can influence the binding of cardiolipin.46 The molecular structure of β2GPI has been elucidated,47,48 and this has given insight into the way it binds to phospholipids via epitopes. The structure of β2GPI, which consists of five domains, has been examined in order to identify which contain epitopes that bind phospholipids. Domain V is not a major binding site in view of its hydrophobic loop that prevents binding to positively charged phospholipids.49,50 Current evidence points to domain I as the main binding site for phospholipid in patients with the syndrome,51–53 and that a mutation of arginine to glycine at position 43 ameliorated the binding in patients with disease.54
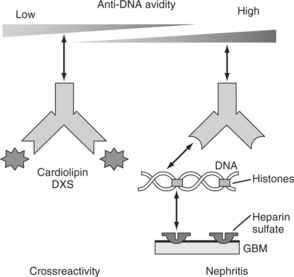
How antiphospholipid antibodies actually promote thrombosis is not fully understood, but they may in some way interfere with the endothelial cells, thereby reducing production of prostacyclin; they may also affect other fibrinolytic mechanisms. Again, this may be mediated by binding to heparan sulfate (Fig. 16.2).
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


