Fig. 6.1
Mechanism of exon skipping therapy in DMD and DYSF. The deletion of exon 52 in the DMD gene results in an out-of-frame transcript that does not produce functional protein (upper left). Antisense oligonucleotides are able to bind in a sequence-specific manner to regions of pre-mRNA and modulate splicing through interference with the spliceosome, resulting in the in-frame skipping of exon 51 and restoration of the reading frame (upper right). A nonsense mutation in exon 32 of the DYSF gene generates a novel stop codon, disrupting the reading frame and preventing the production of functional protein (bottom left). Antisense oligonucleotides can bind to a region of exon 32 and prevent incorporation of the exonic sequence into the final mRNA transcript, restoring the reading frame and producing a truncated protein that may retain some functionality (bottom right). Phasing or “framed-ness” of each exon is denoted by the shape of the ends of the exon – ends that fit together are in-frame
One possible explanation for the remarkable success in antisense-based therapies for the treatment of DMD lies in understanding the pathology of the disease and the changes which take place at the cellular level. Without dystrophin protein, the structural integrity of the sarcolemma is severely compromised, resulting in small perforations in the cell membrane [44]. These perforations result in a “leaky” cell membrane and could help facilitate easier transport of AOs into the muscle fiber [18]. This would result in the increased likelihood that AOs will be able to reach their intracellular targets. As discussed previously, cell delivery has always posed a major hurdle to nucleic acid therapies – but in the case of DMD, it may be that the very nature of the disease makes it more amenable to this particular kind of therapeutic approach.
6.4.2 Fukuyama Congenital Muscular Dystrophy
Fukuyama congenital muscular dystrophy (FCMD) is an autosomal recessive inherited form of muscular dystrophy more commonly associated with the Japanese population (about 1 in 10,000 births) [45, 46]. The FCMD gene encodes the protein fukutin, a putative glycosyltransferase [46–48]. Although its exact function is not fully understood, fukutin is a ubiquitously expressed protein (although expressed at much higher levels in skeletal muscle) thought to glycosylate cell-surface glycolipids or glycoproteins, including α-dystroglycan, a member of the dystrophin-associated glycoprotein complex (DGC) [47]. Patients with FCMD harbor a 3-kb retrotransposon insertion in the 3′-untranslated region (UTR) of the gene, which introduces a splicing error that causes premature truncation of exon 10 [28]. Researchers have successfully utilized a cocktail of multiple vPMOs targeting intronic and exonic splice enhancers to restore normal fukutin expression and protein levels both in a mouse model and in human patient cells (Fig. 6.2) [28]. Although initially promising, there have yet to be any further investigations into the antisense-mediated treatment of FCMD.
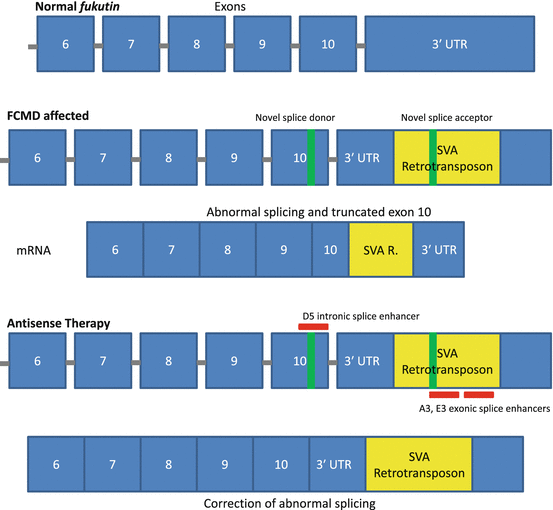
Fig. 6.2
Mechanism of antisense therapy in treating FCMD. The addition of a retrotransposal unit into the 3′ UTR of the fukutin gene generates novel splice acceptor and donor sites, resulting in aberrant splicing and truncation of exon 10. A cocktail of three antisense oligos targeting splice enhancer regions is able to correct splicing abnormalities and restore protein expression
6.4.3 Dysferlinopathy
Mutations in the dysferlin (DYSF) gene are associated with three autosomal recessive inherited muscular dystrophies – limb-girdle muscular dystrophy type 2B (LGMD2B), Miyoshi myopathy (MM), and distal myopathy with anterior tibial onset (DMAT) – and are collectively referred to as the dysferlinopathies [49–56]. The clinical symptoms of dysferlinopathy present across a wide pathological spectrum which narrows with disease progression until each subtype becomes practically indistinguishable. The protein product of DYSF, dysferlin, is a ubiquitously expressed transmembrane protein found at higher levels in skeletal and cardiac muscles where it plays an essential role in vesicle trafficking and plasma membrane resealing [54]. Providing “natural” proof of principle evidence that exon skipping could be amenable to treating dysferlinopathies, a female patient was previously reported as having a DYSF mutation causing the in-frame skipping of exon 32. Although the patient had two daughters with homozygous null mutations and severe LGMD2B, the patient herself presented with only mild symptoms, a phenotype attributed to the action of truncated DYSF protein caused by her mutation [57]. These observations drove subsequent investigations into the feasibility of DYSF exon skipping in vitro. One group utilized myoblasts generated from dysferlinopathy patients to test the effectiveness of 2′-O-methyl AOs targeting exonic splice enhancer/silencer sites and was able to achieve efficient exon skipping of exon 32 (Fig. 6.1) [58]. Another natural observation supporting the potential therapeutic action of truncated dysferlin comes from the report of a male dysferlinopathy patient who, despite proximodistal muscle weakness, was still ambulant unassisted at 41 years [59]. Intramuscular injections of a viral vector containing a DYSF construct corresponding to the patient’s mutation were administered to DYSF-null mice and resulted in significant protein expression and restoration of membrane resealing ability [59]. As yet, no in vivo investigations have been reported that assess the effectiveness of AOs in restoring membrane resealing ability and ameliorating dysferlinopathy phenotype, although several promising exonic targets have been identified and are under investigation for their amenability to antisense-mediated exon skipping based on in-frame translational shifts, confirmed redundancy, and their disassociation with any reported pathology [60].
6.4.4 Myotonic Dystrophy
Myotonic dystrophy type 1 (DM1) is a multisystemic neuromuscular disease characterized by progressive muscle wasting, myotonia, cardiac conduction deficits, mental retardation, insulin resistance, and cataracts, although the combination of these symptoms and their severity varies from patient to patient [61]. DM1 is caused by an expanded CTG tract in the 3′ UTR of the dystrophia myotonica-protein kinase (DMPK) gene, which is believed to result in RNA-gain-of-function toxicity [62]. A hallmark feature of DM pathogenesis is aberrant splicing (spliceopathy). DM spliceopathy is believed to occur due to protein binding with expanded repeat DMPK transcripts, and these spliceopathies are thought to be responsible for many DM symptoms, with more than 30 spliceopathic genes having been identified in DM patients [63–68]. Some spliceopathic genes which have been implicated in DM pathology, specifically in the progression of myotonia, include the chloride channel 1 (CLCN1) and muscleblind-like splicing regulator 1 (MBNL1) genes [69–71]. Antisense-mediated degradation of mutant DMPK transcripts and/or the correction of abnormal splicing in DM-related spliceopathic genes through nucleic acid-based therapy are, therefore, promising avenues for treating DM1 (Fig. 6.3). Using 2′-O-methyl AOs, a novel DM1 myoblast-myotube cell model, and a well-characterized DM mouse model (HSALR), Mulders et al. achieved efficient silencing of mutant DMPK transcripts in vitro and in vivo, a reduction in the amount of RNA nuclear foci, and normalization of spliceopathy in several affected genes [72]. In human cells, expanded CUG transcripts were significantly reduced following the addition of AOs, and in DM300-328-XXL mice harboring a 45-kb human genomic fragment containing human DMPK with an expanded CTG repeat, there were reduced levels of mutant transcripts following AO injection into skeletal muscles [73]. In another study, systemically administered 2′-MOE AOs were effective in reducing expanded CUG repeats and nuclear foci and achieved transcriptome normalization and normalization of splicing in four genes: sarcoplasmic/endoplasmic reticulum calcium ATPase 1 (ATP2A1 or SERCA1), titin (TTN), LIM domain binding 3 (LDB3 or ZASP), and chloride channel 1 (CLCN1) with no signs of toxicity [74]. In the same study, persistent AO activity was detected up to 1 year following initial treatment and was accompanied by the sustained amelioration of several myopathic features including reduced numbers of centrally nucleated fibers and increased muscle fiber diameter [74]. Focusing on the correction of splicing in CLCN1, researchers employed PMOs to prevent incorporation of frame-shift-causing exon 7a and were able to suppress exon 7a inclusion, restore CLCN1 protein expression, rescue channel function, and reverse myotonia using two different DM mouse models [75]. Another group achieved amelioration of myotonia in mice who received PMOs delivered via bubble liposome-ultrasound, highlighting an effective method of AO administration and further demonstrating the feasibility of AO-based therapies for the treatment of DM [64].
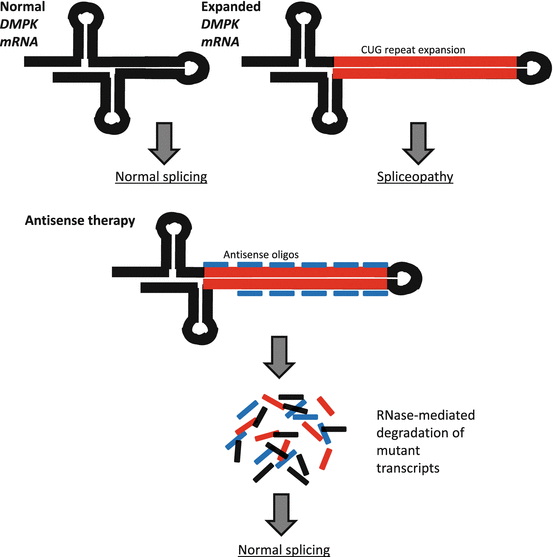
Fig. 6.3
Antisense-mediated degradation of toxic DMPK expanded repeat transcripts. DM1 is caused by a CUG repeat expansion in the DMPK gene, which results in spliceopathy in several other genes. Expression of mutant DMPK transcripts can be silenced through the use of antisense oligos which catalyze RNase-mediated degradation
6.5 Current Investigations and Clinical Trials
The road toward implementing AOs in the clinical arena has not been an easy one. Despite years of study, AOs as a licensed therapeutic agent have had very little success in reaching the market. To date, only two antisense drugs have ever been approved by the United States Food and Drug Administration: Vitravene (Isis Pharmaceuticals, Carlsbad, CA, USA), for the treatment of cytomegalovirus retinitis in HIV-positive AIDS patients, and Kynamro® (Isis Pharmaceuticals, Carlsbad, CA, USA), for the treatment of familial hypercholesterolemia [3]. Owing to recent and ongoing advancements in AO chemistries, several major hurdles facing nucleic acid-based therapies have been overcome. Some major hurdles remain, such as unknown long-term safety following AO administration, the limited efficacy of AOs in cardiac muscle, and limited applicability (only about 13 % of DMD patients are amenable to exon 51 skipping) [76, 77]. As research continues in this field, and it is already advancing at a rapid pace, it may not be long before an antisense drug for the treatment of muscular dystrophy comes to the clinic.
Based on the status of current clinical trials, the most promising candidate for an antisense drug is likely to be an AO designed for the treatment of Duchenne muscular dystrophy. To date, no other form of muscular dystrophy has received as much exposure in the preclinical and clinical trial world as that of DMD. These clinical trials have centered exclusively on single exon skipping, with exon 51 being one of the most promising targets [24, 26, 78–81], although clinical trials involving exons 44, 45, and 53 single exon skipping are also ongoing [2].
One of the leading developers of nucleic acid-based therapeutics is Sarepta Therapeutics. Their lead antisense drug, PMO-based Eteplirsen, is currently the focus of phase II/III clinical trials aimed at skipping exon 51 in the dystrophin gene for treating DMD. According to a recent report from a phase IIb trial, Eteplirsen significantly improved pulmonary function in patients treated through 120 weeks. This finding comes in the wake of preceding data from a 120-week study which found a significant improvement in walking ability in treated patients, as demonstrated by the 6-min walk test. No clinically significant negative effects due to Eteplirsen treatment have been reported. Another DMD exon 51-targeting drug, the 2′OMePS oligo Drisapersen, was developed jointly by GlaxoSmithKline (GSK) and Prosensa, and previous phase II clinical trial data demonstrated the safety, tolerability, and effectiveness of the drug at improving walking distance after 12 weeks of treatment. Unfortunately, Drisapersen did not demonstrate significant improvement in the 6-min walk test in a more recent phase III clinical trial. As a result, GSK is no longer involved in pursuing further clinical trials with Drisapersen, although Prosensa continues to be involved with the drug’s development. There are plenty of lessons we can learn from this failure as recently pointed out [3]. For example, the doses of injections in this trial (up to 6 mg/kg) were much lower than the effective dose in mdx mice (2 mg/mouse; approximately 75–100 mg/kg) [41]. A very recent development in DMD clinical trials involves the joint efforts of Nippon Shinyaku Co., Ltd. and the National Center of Neurology and Psychiatry (NCNP, Kodaira City, Japan), which have been developing a PMO-based antisense drug that facilitates exon 53 skipping in DMD patients. The first clinical trial involving their lead candidate drug NS-065/NCNP-01 is underway in 2014. This marks the first ever nucleic acid-based clinical trial in Japan and the first DMD exon 53-targeting clinical trial in the world.
While single exon skipping may be a focus of current clinical trials, future investigations involving multiple exon skipping could widen the door of applicability for AOs in treating muscular dystrophies, especially DMD. The majority of DMD patients (about 63 %) with deletion mutations harbor a mutation between the region of exons 45–55 in the DMD gene, a region known as the “mutation hot spot” [27, 37]. The development of a novel antisense approach to skipping exons 45–55 in the human DMD gene could overcome the enormous clinical heterogeneity observed in DMD by providing a single therapeutic tactic to treating a large proportion of patients. Furthermore, naturally occurring exons 45–55 deletions in patients are associated with an exceptionally mild, even asymptomatic phenotype [27, 35]. Bodywide exon skipping of exons 45–55 has been accomplished in a mouse model of DMD but has yet to be reported in human cells [27].
With successes in recent clinical trials, ongoing translational research using both in vitro and in vivo models, and continual advancements in AO chemistries, nucleic acid therapies for muscular dystrophies have become one of the most rapidly improving therapeutic strategies in medical research and may soon cease to be a technology exclusive to just the lab bench or to clinical trial cohorts.
Acknowledgments
This work was supported by the University of Alberta Faculty of Medicine and Dentistry, Parent Project Muscular Dystrophy (USA), The Friends of Garrett Cumming Research Funds, HM Toupin Neurological Science Research Funds, Muscular Dystrophy Canada, Canada Foundation for Innovation, Alberta Enterprise and Advanced Education, Jesse’s Journey, Slipchuk SMA Research Funds, the Women and Children’s Health Research Institute, and Canadian Institutes of Health Research.
References
1.
2.
Lee J, Yokota T (2013) Antisense therapy in neurology. J Pers Med 3(3):144–176. doi:10.3390/jpm3030144#sthash.EzKjmG9w.dpuf PubMedCentralCrossRefPubMed
3.
Touznik A, Lee JJ, Yokota T (2014) New developments in exon skipping and splice modulation therapies for neuromuscular diseases. Expert Opin Biol Ther 14(6):809–819. doi:10.1517/14712598.2014.896335 CrossRefPubMed
4.
Spiegelman WG, Reichardt LF, Yaniv M, Heinemann SF, Kaiser AD, Eisen H (1972) Bidirectional transcription and the regulation of Phage lambda repressor synthesis. Proc Natl Acad Sci U S A 69(11):3156–3160PubMedCentralCrossRefPubMed
5.
Green PJ, Pines O, Inouye M (1986) The role of antisense RNA in gene regulation. Annu Rev Biochem 55:569–597. doi:10.1146/annurev.bi.55.070186.003033 CrossRefPubMed
6.
Itoh T, Tomizawa J (1980) Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proc Natl Acad Sci U S A 77(5):2450–2454PubMedCentralCrossRefPubMed
7.
Simons RW, Kleckner N (1988) Biological regulation by antisense RNA in prokaryotes. Annu Rev Genet 22:567–600. doi:10.1146/annurev.ge.22.120188.003031 CrossRefPubMed
8.
Tomizawa J, Itoh T, Selzer G, Som T (1981) Inhibition of ColE1 RNA primer formation by a plasmid-specified small RNA. Proc Natl Acad Sci U S A 78(3):1421–1425PubMedCentralCrossRefPubMed
9.
Light J, Molin S (1983) Post-transcriptional control of expression of the repA gene of plasmid R1 mediated by a small RNA molecule. EMBO J 2(1):93–98PubMedCentralPubMed
10.
Mizuno T, Chou MY, Inouye M (1984) A unique mechanism regulating gene expression: translational inhibition by a complementary RNA transcript (microRNA). Proc Natl Acad Sci U S A 81(7):1966–1970PubMedCentralCrossRefPubMed
11.
12.
Zamecnik PC, Stephenson ML (1978) Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci U S A 75(1):280–284PubMedCentralCrossRefPubMed
Related posts:
 Fukuyama Congenital Muscular Dystrophy: Clinical Aspects
Targeting the Type I TGF-β Receptor for Treating Caveolin-3-Deficient Autosomal Dominant Limb-Girdle Muscular Dystrophy Type 1C and Muscle Wasting Disorders
Therapeutic Approach of iPS Cell Technology for Treating Muscular Dystrophy
Myotonic Dystrophy
Translational Research on DMD in Japan
Patient Registries for International Harmonized Clinical Development
Fukuyama Congenital Muscular Dystrophy: Clinical Aspects
Targeting the Type I TGF-β Receptor for Treating Caveolin-3-Deficient Autosomal Dominant Limb-Girdle Muscular Dystrophy Type 1C and Muscle Wasting Disorders
Therapeutic Approach of iPS Cell Technology for Treating Muscular Dystrophy
Myotonic Dystrophy
Translational Research on DMD in Japan
Patient Registries for International Harmonized Clinical Development
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


