Concussion, or mild traumatic brain injury (TBI), affects millions of patients worldwide. Understanding the pathophysiology of TBI can help manage its repercussions. The brain is significantly altered immediately following mild TBI because of metabolic, hemodynamic, structural, and electrophysiologic changes. This process affects cognition and behavior and can leave the brain vulnerable for worse injury in the setting of repeat insult. This article is an update of our previously published review, reporting relevant and current studies from the bench to the bedside of mild TBI. Understanding the pathobiology can help prevent and treat mild TBI.
Key points
- •
Cerebral metabolism is altered immediately after concussion and may increase vulnerability for an interval after injury.
- •
Advanced neuroimaging, such as functional MRI, MR spectroscopy and diffusion tensor imaging can demonstrate alterations associated with concussive brain injury.
- •
Cumulative concussive brain injury has been associated with increased neurocognitive symtpoms.
- •
Pituitary dysfunction has been reported after chronic repetitive concussions and can affect quality of life.
Introduction
Concussion (mild traumatic brain injury [TBI]), particularly with organized sports, has come to the forefront of popular culture. Affecting about 1.6 million to 3.8 million athletes a year, its short-term and chronic effects have been increasingly recognized as notable sports-related injuries. Thanks to widely disseminated checklist-based assessment tools and greater symptom awareness in the sports community, it is being diagnosed more frequently in amateur and professional venues alike. Public media have also embraced this condition, chiding collegiate coaches who ignore concussion and producing feature films depicting its chronic effects.
Nearly 20 years ago, the American Association of Neurology (AAN) introduced their grading system for the diagnosis and treatment of concussions. These guidelines were focused on helping clinicians counsel patients to manage their symptoms and return to play. It has since become apparent that it is difficult to predict recovery based on initial concussion severity. The AAN released updated guidelines in 2013, based on a systematic, evidence-based review of the available literature. These guidelines underscored the importance of early recognition of mild TBI to address symptoms and potentially prevent more severe injury. Early assessment tools, such as the Sports Concussion Assessment Tool, are endorsed for sideline nonphysicians with a high positive predictive value for concussion identification. However, much like in consensus statements by the Concussion in Sport Group (introduced in 2001 and thrice updated), these guidelines eliminated a severity grading system and, instead focused on factors affecting the timing of an athlete’s return to play. This approach is primarily based on the presence and severity of general/neurologic symptoms or demonstrable neuropsychological impairments, as well as an individualized assessment of risk factors for prolonged recovery, such as history of prior concussions; younger age; symptoms of migraine headache, fogginess, and dizziness; learning disability or attention deficit; on-field mental status change; and possibly psychiatric comorbidities such as depression and anxiety.
Experimental animal models have been central to understanding the pathophysiology of concussive brain injury. Typically performed with rodents, techniques include closed-skull weight drop, closed-skull controlled impact, and lateral fluid percussion injury (FPI). Through these models, researchers can glean clinically relevant mechanistic insight and are able to better characterize molecular alterations, ionic and neurotransmitter disturbances, synaptic perturbations, and microstructural changes. More recently, high-resolution MRI, diffusion tensor imaging (DTI)/tractography, functional MRI, and magnetic resonance (MR) spectroscopy have allowed real-time imaging of structural and molecular changes without sacrificing the animal. An added benefit is the cross-availability of these imaging modalities for human data acquisition. Such translational capacity has shown utility for bench-to-bedside research. In particular, DTI and functional MRI have been shown to be reliable research markers of mild TBI-induced injury; identifying neuronal damage or dysfunction in the setting of otherwise normal macrostructural imaging (ie, computed tomography scanning; T1/T2 MRI). Other, more invasive techniques include microdialysis of the injured brain and its histopathologic evaluation.
This article updates our previously published article with additional focus on metabolic vulnerability to repeat injury, advanced MRI modalities assessing postconcussive outcomes, chronic changes following repetitive concussive injury, and pituitary dysfunction following prolonged exposure to mild TBI.
Introduction
Concussion (mild traumatic brain injury [TBI]), particularly with organized sports, has come to the forefront of popular culture. Affecting about 1.6 million to 3.8 million athletes a year, its short-term and chronic effects have been increasingly recognized as notable sports-related injuries. Thanks to widely disseminated checklist-based assessment tools and greater symptom awareness in the sports community, it is being diagnosed more frequently in amateur and professional venues alike. Public media have also embraced this condition, chiding collegiate coaches who ignore concussion and producing feature films depicting its chronic effects.
Nearly 20 years ago, the American Association of Neurology (AAN) introduced their grading system for the diagnosis and treatment of concussions. These guidelines were focused on helping clinicians counsel patients to manage their symptoms and return to play. It has since become apparent that it is difficult to predict recovery based on initial concussion severity. The AAN released updated guidelines in 2013, based on a systematic, evidence-based review of the available literature. These guidelines underscored the importance of early recognition of mild TBI to address symptoms and potentially prevent more severe injury. Early assessment tools, such as the Sports Concussion Assessment Tool, are endorsed for sideline nonphysicians with a high positive predictive value for concussion identification. However, much like in consensus statements by the Concussion in Sport Group (introduced in 2001 and thrice updated), these guidelines eliminated a severity grading system and, instead focused on factors affecting the timing of an athlete’s return to play. This approach is primarily based on the presence and severity of general/neurologic symptoms or demonstrable neuropsychological impairments, as well as an individualized assessment of risk factors for prolonged recovery, such as history of prior concussions; younger age; symptoms of migraine headache, fogginess, and dizziness; learning disability or attention deficit; on-field mental status change; and possibly psychiatric comorbidities such as depression and anxiety.
Experimental animal models have been central to understanding the pathophysiology of concussive brain injury. Typically performed with rodents, techniques include closed-skull weight drop, closed-skull controlled impact, and lateral fluid percussion injury (FPI). Through these models, researchers can glean clinically relevant mechanistic insight and are able to better characterize molecular alterations, ionic and neurotransmitter disturbances, synaptic perturbations, and microstructural changes. More recently, high-resolution MRI, diffusion tensor imaging (DTI)/tractography, functional MRI, and magnetic resonance (MR) spectroscopy have allowed real-time imaging of structural and molecular changes without sacrificing the animal. An added benefit is the cross-availability of these imaging modalities for human data acquisition. Such translational capacity has shown utility for bench-to-bedside research. In particular, DTI and functional MRI have been shown to be reliable research markers of mild TBI-induced injury; identifying neuronal damage or dysfunction in the setting of otherwise normal macrostructural imaging (ie, computed tomography scanning; T1/T2 MRI). Other, more invasive techniques include microdialysis of the injured brain and its histopathologic evaluation.
This article updates our previously published article with additional focus on metabolic vulnerability to repeat injury, advanced MRI modalities assessing postconcussive outcomes, chronic changes following repetitive concussive injury, and pituitary dysfunction following prolonged exposure to mild TBI.
Neurometabolic cascade of concussion
The mechanical trauma exerted on the brain during a concussive event occurs via acceleration and deceleration forces on the neuronal structures. These forces set off a complex cascade of neurochemical and neurometabolic events. A mechanical disruption of cell membranes and axonal stretching causes unregulated efflux of ions through previously regulated ion channels. This rapid depolarization results in indiscriminate release of numerous neurotransmitters; primarily excitatory amino acids such as glutamate. This process potentiates the existing ionic flux to a wider neuronal region. Simultaneously, the Na/K ATP-dependent pump is active to reestablish ionic balance. However, this results in depleted energy stores ( Fig. 1 ). These molecular cascades alone can be the cause of the postconcussive symptoms and may result in subsequent cerebral hypofunction or, rarely, permanent damage. Often, these effects are self-limited and transient, although in the setting of repetitive injury this could result in more prolonged neurologic deficits. These molecular cascades are discussed in further detail later.
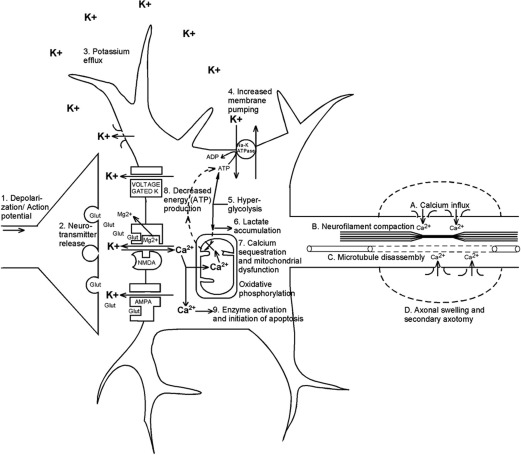
Glutamate release and ionic flux
The deformation of neuronal membranes after biomechanical injury results in excessive potassium efflux into the extracellular space. Simultaneously, there is unregulated release of excitatory amino acids (EAAs), especially glutamate, which binds to the kainate, N -methyl d -aspartate (NMDA), and d -amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid ionic channels. This process causes further regional depolarization, predominantly mediated through NMDA receptors (NMDAR); tetrodotoxin has little effect on this cascade and kynurenic acid (an NMDAR antagonist) attenuates it. Hence, a widespread relative suppression of neurons occurs, creating a condition resembling spreading depression. This concept of spreading depression has been suggested as a mechanism not only for migraine pathophysiology but also for seizures and, more recently, with secondary injury after traumatic brain injury (TBI).
The ATP-dependent Na + /K + pumps are active to maintain or normalize membrane potentials, requiring high levels of glucose metabolism, primarily through aerobic respiration. After TBI, the pump activity is maximal and quickly reduces intracellular glucose stores. As seen in experimental TBI in rats, the increased glucose metabolism occurs immediately after injury and may last from 30 minutes to 4 hours. Simultaneously, there is inefficient oxidative metabolism, likely from mitochondrial dysfunction. Hence, primarily anaerobic metabolism of glucose occurs with rampant accumulation of lactate, also seen in the extracellular space. There is a resultant local acidosis, increased membrane permeability, and cerebral edema. Note that lactate may be used as an energy source by neurons, once mitochondrial function resumes. Lactate administration has been used as a treatment option for patients with severe TBI with possible positive cerebral metabolic and hemodynamic effects.
Glucose metabolism and mitochondrial effects
Following concussive injury, 2 major alterations of glucose metabolism have been described: hyperglycolysis and oxidative dysfunction. Local cerebral metabolic rates for glucose are increased within the first 30 minutes following lateral FPI, up to 30% to 46% greater than control levels. After 6 hours, there is a relative glucose hypometabolism (approximately 50%, depending on brain region) that can last up to 5 to 10 days, depending on age, injury model, and severity. A similar profile of hyperglycolysis followed by glucose hypometabolism has been reported based on fluorodeoxyglucose (FDG)-PET measurements after human TBI. The duration of late hypometabolism may be months after moderate to severe TBI. More recently, prolonged postconcussive (mild TBI) regional hypometabolism has been shown based on FDG-PET imaging compared with age-matched controls, and has been suggested as a possible substrate of postconcussive symptoms in these patients. However, this has not yet been studied with longitudinal within-subject FDG-PET analysis.
Oxidative (mitochondrial) dysfunction in the acute phase following TBI is thought to be a result of the significant influx of Ca ++ through open NMDA channels and voltage-sensitive Ca channels. There may be resultant calcium accumulation in the mitochondria. This calcium accumulation is reflected by decreased metabolic biomarkers such as ATP/ADP ratio, NADH/NAD + ratio, and N -acetyl aspartate (NAA) seen after mild TBI in rat models, with lowest nadirs 3 days following injury. Mitochondrial oxidative function is thought to be diminished up to 10 days following injury, partly based on downregulated cytochrome oxidase activity after FPI assessed by histochemistry.
Alternative energy sources are available to the healthy and postconcussive brain. These energy sources include creatine (Cr), creatine-phosphate, and ketone bodies. Recent studies suggest that Cr and Cr-phosphate are not viable fuel sources following injury, based on decreased levels along with NAA, ATP/ADP ratio, and phosphatidyl choline following mild TBI in rats. This finding was reflected in concussed athletes with decreased NAA/Cr ratios on MR spectroscopy.
Ketone bodies have been known to be an alternative fuel source for the body during times of stress or starvation. Animal data suggest that although glucose metabolism is altered immediately following concussive TBI, glucose may not be the best fuel for the injured brain. Ketosis or a ketogenic diet in rats following cortical contusion injury showed age-dependent decreased glucose metabolism. Subsequent studies suggest that ketogenic diet is associated with smaller cerebral contusion volumes and improved behavioral outcomes. Although the neuroprotective aspects of ketosis have yet to be systematically studied after mild TBI, it is suggested that ketosis induced by fasting may be most applicable in the first 24 hours following moderate, but not severe TBI. The neuroprotective implications of ketosis have yet to be investigated systematically after mild TBI.
Repeat TBI during this period of cellular ionic disturbance, decreased cerebral blood flow, and glucose metabolic dysfunction is thought to result in more severe brain injury, described clinically as second impact syndrome. However, definitive description of this clinical entity has been controversial, and the role of glucose hypometabolism on brain injury has not yet been determined to be protective or exacerbating after a second insult.
Cerebral blood flow
Severe TBI has been associated with cerebral blood flow (CBF) perturbations, characterized by a triphasic response. This condition has been well characterized, but with some variability in the degree of post-TBI ischemia. Martin and colleagues reported a series of 125 patients, assessed by cerebral arteriovenous delivery of oxygen, cerebral metabolic oxygen consumption, and vasospasm (measured by transcranial Doppler). Cerebral hypoperfusion (mean CBF, 32.3 mL/100 g/min) was noted at postinjury day (PID) 0, followed by cerebral hyperemia (mean CBF, 46.8 mL/100 g/min) and increased middle cerebral artery (MCA) velocities (86 cm/s) at PID 1 to 3. Subsequently, there is a period of cerebral vasospasm with decreased CBF of 35.7 mL/100 g/min and increased MCA velocities (96.7 cm/s) at PID 4 to 15.
Decreased postinjury CBF has been shown in patients with mild TBI as well. Maugans and colleagues reported a small series of pediatric patients with concussion who were evaluated with MRI CBF assessment (phase contrast quantitative flow angiography). There was a notable decrease in CBF relative to case-matched controls at initial evaluation (within 72 hours of injury: mean, 44.75 hours) of 38 ± 13.4 mL/100 g/min versus 48 ± 9.8 mL/100 g/min. Only 3 patients (27%) had recovery of CBF at 14 days, with 7 patients (67%) similarly at final follow-up (mean 41 days). There was no direct correlation with CBF rates and immediate post-concussion assessment and cognitive test (ImPACT) total symptom scores (TSS), despite increased TSS at the initial and 14-day assessments that resolved at final assessment.
These findings were supported in a recent longitudinal adult concussion athlete study by Meier and colleagues, showing decreased regional CBF at 1 day and 1 week following injury relative to controls, which recovered by 1 month. There was a correlation with decreased midinsular CBF at 1 month in patients who were slower to recover. This effect has been shown in the chronic phase as well, with decreased CBF reported in symptomatic patients at 3 to 12 months following concussion. The triphasic response has not been assessed in mild TBI, primarily because of the inability to reliably assess CBF data immediately after injury.
Perilesional cerebral edema assessed by MRI has been reported in animal models (cortical contusion) for severe TBI. This effect is maximal through 4 days following injury, which then recovers over the following 2 weeks correlated with the neuroscore (a behavioral scale of neurologic function). Although cerebral edema was not seen in mild TBI animal models (FPI), there is mild increase in the apparent diffusion coefficient with concomitant functional MRI impairment at 1 and 7 days following weight-drop injury.
Axonal injury and diffusion tensor imaging
In severe TBI, typically from blunt injury, diffuse axonal injury (traumatic axonal injury) has been well described. This injury is thought to be caused by the mechanical deformation of the axonal cell membranes resulting not only in the aforementioned metabolic and ionic changes but also in neurofilament compaction. In the acute postinjury phase (5 minutes to 6 hours), this can occur by either phosphorylation or calpain-mediated proteolysis of sidearms. Subsequently, the calcium influx can also destabilize the microtubules. There is resultant dysfunction of axonal transport, resulting in axonal blebbing and eventual disconnection.
This phenomenon may also occur after mild TBI, albeit with less severity and potentially reversible effects. A murine model study showed damage mainly at the axonal level, with minimal effect to the neuronal cell bodies or myelin sheaths following mild TBI via FPI. This damage progressed across various cortical and subcortical structures over 4 to 6 weeks and correlated with impaired spatial learning and memory deficits (Morris water maze [MWM]). Other in vivo and in vitro models of axonal stretch show similar damage, with impaired axonal transport, axonal undulations, blebbing, and disconnection.
MRI DTI and tractography are recent advances in modern imaging with significant clinical research applications. Fractional anisotropy (FA) is a measure of linear water diffusion that decreases when directionality of white matter tracts is disturbed, such as after axonal disconnection or damage to myelin sheaths Higher FA values correlate with more linear water diffusion (ie, along tubes such as axonal tracts). Postinjury alterations in FA have been hypothesized to be related to transient axonal swelling. This condition has been seen in pediatric, adolescent, and adult patients following mild TBI/concussion and, in some cases, has been correlated with cognitive dysfunction.
In a pediatric study of mild and moderate TBI, FA was shown to be decreased in white matter subcortical regions (inferior frontal, superior frontal, and supracallosal), but unchanged in the corpus callosum. There was a correlation of motor speed, executive function, and behavioral ratings with these FA values. Similar structural perturbations have also been seen chronically after mild TBI in adults, affecting regions such as the genu of the corpus callosum, the cingulum, the anterior corona radiata, and the uncinate fasciculus ( Fig. 2 ). Although there was a direct correlation between the decrease in FA and cognitive function, there was no association with the number of microhemorrhages seen on high-resolution MRI. In contrast, a study in adolescent patients with mild TBI showed increased in the corpus callosum FA levels seen early (6 days) following injury (thought to be caused by axonal edema rather than disruption). However, these findings did correlate with postconcussive symptoms confirmed by cognitive, affective, and somatic scores on the Rivermead Post-Concussion Symptoms Questionnaire and Brief Symptom Inventory. This increase in FA was supported by Mayer and colleagues, who reported a study of 15 pediatric patients with mild TBI with increased FA scores in the corpus callosum, cingulum, internal capsule, and cerebral peduncles.
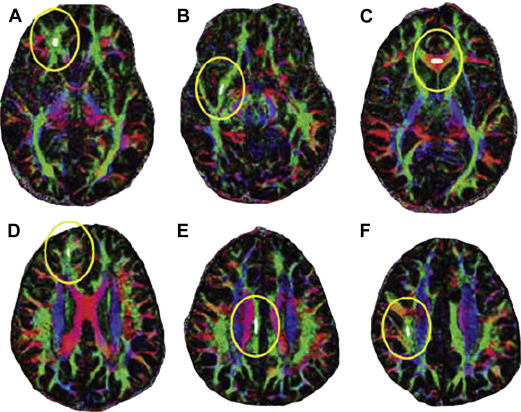
More recently, corpus callosum function has been assessed after pediatric moderate to severe TBI by interhemispheric transfer time (IHTT) using event-related potentials. There is notable variability of IHTT within sample populations, although slower IHTT has been associated with decreased performance scores. Dennis and colleagues reported in the slow-IHTT group a notable decrease in FA values across select axonal bundles ( Fig. 3 ). In contrast, in the normal-IHTT group, there was no significant difference in FA values compared with controls. Hence, such studies support the correlation of lower FA values and impaired cognitive function via diminished connectivity. There also seems to be a correlation with specific white matter structures and select cognitive or behavioral deficits. These studies suggest that DTI assessing axonal injury is a sensitive and powerful measure of the effects from TBI of all severities.
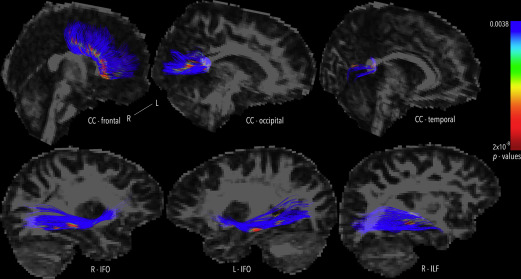
Altered brain activation
The NMDA glutamate receptor is of particular interest with regard to neuronal activity and calcium regulation following TBI. This tetrameric channel (2 NR1 and 2 NR2 subunits) requires 2 signals to be activated: membrane voltage change and glutamate binding. This activation releases an Mg++ ion within its working channel and allows calcium flux into the neuron. Developmentally, there is a shift of the channel composition in the rat brain, with downregulation of NR2B (slower channels) and upregulation of NR2A (faster channels) with maturity.
Following lateral FPI in pediatric (postnatal day 19) rats, the relative expression of the NR2A subunit is downregulated by PID 4 with apparent recovery by PID 7. The expression of NR2B and NR1 subunits remains unchanged, suggesting a possible intrinsic neuroprotective mechanism of calcium ion regulation following TBI. Long-term potentiation (LTP) and long-term depression, functions of learning and memory, are associated with NMDA channel activity and composition. As expected, there is impaired LTP following experimental TBI, at PID 2, with apparent partial recovery by PID 7 to 15. However, maintenance of LTP is deficient up to 8 weeks postinjury.
Postconcussive patients show abnormal activation of neural circuits, associated with cognitive and behavioral deficits. Blood oxygen level–dependent sequences obtained in functional MRI before and after cognitive tasks show a hyperactivation in the postconcussive brain at 1 week ( Fig. 4 ). When abnormal activation is seen following concussive brain injury, these athletes seem to have a more prolonged clinical recovery. With task-based assessment, increased recruitment of cognitive resources has been seen, correlated with concussion severity.
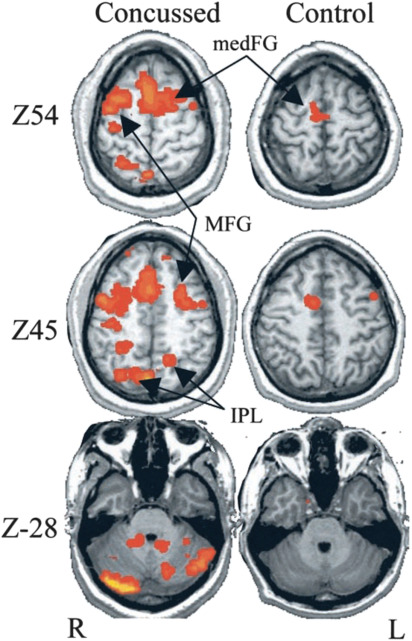
Acute responses to repeat concussion
Although the postconcussive symptoms are important to identify and monitor, another potential reason to limit return to activity is the second impact syndrome following repeat concussive injury of the vulnerable brain. Severe examples include patients with catastrophic cerebral edema following apparent mild TBI/concussion resulting in coma, severe neurologic deficits, and even death.
Although predictive factors and the time interval for return to activity following concussion remain debated, it is evident that the cerebral physiology is altered following mild TBI, which may render the brain less functional and more vulnerable. This period of vulnerability, characterized by the aforementioned metabolic, hemodynamic, structural, and electrophysiologic dysfunction, can increase the vulnerability of the brain to repeat injury. This window of impairment has been characterized by animal studies and supported by clinical experience.
A pair of recent studies attempted to assess the vulnerability interval in mice. Meehan and colleagues assessed daily, weekly, and monthly repetitive concussion intervals with regard to cognitive function assessed by MWM performance. Mice sustaining 5 concussions at daily or weekly intervals had similar decreased cognitive function compared with controls. However, those sustaining 5 concussions at monthly intervals had no difference from controls. Those same mice had similar dysfunction when subsequently concussed daily. These cognitive tests suggested that concussed brain is vulnerable at up to 1 week following injury. Mannix and colleagues published a follow-up long-term study showing that mice that sustained biweekly and monthly injuries did not have significant cognitive deficits. Daily and weekly concussed animals had evidence of histologic changes associated with chronic cognitive dysfunction (astrocytosis). These animal studies place the time window of vulnerability between 1 and 2 weeks following injury.
In a rat weight-drop (mild TBI) experiment by Vagnozzi and colleagues, NAA, ATP, and the ATP/ADP ratio were shown to be significantly decreased at PID 2 following repeat concussion. Peak deficits were seen when repeat injuries were separated by a 3-day interval. At this combination, metabolic perturbation was similar to a single severe TBI injury. These abnormalities persisted as late as 7 days after double impact, shown in a follow-up study by the same group. Adding to this, Prins and colleagues assessed glucose metabolism (cerebral metabolic rate of glucose) following mild TBI in rat pups. They identified decreased glucose metabolism at PID 1 by 19%. This decrease recovered by PID 3. However, if a second injury was induced during the vulnerable period on PID 1, glucose metabolism was further depressed (36.5%) without recovery at 3 days (25% depressed metabolism). This effect was not seen when repeat injury was performed at PID 6, and correlated with working memory impairments on behavioral testing.
MR spectroscopy has proved to be a good research tool for estimating regional metabolite concentrations in athletes and other individuals with concussion. In one study, 13 athletes who sustained concussions underwent 3-T MR spectroscopy at various postinjury time points. At 3 days postinjury, the NAA/Cr ratio was diminished by 18.5% compared with age-matched control patients (2.2 vs 1.8; P <.05). There was slight improvement at 15 days (1.88) and normalization at 30 days postinjury. Perhaps most interesting are the 3 patients who sustained a repeat concussion between 3 and 15 days after their initial injuries. These patients had a similar initial decrease in their NAA/CA ratios (1.78), but had further decrease at 15 days (1.72), rather than a partial resolution, which took 45 days to recover to baseline. The patients with single concussion reported no symptoms at the time of the 3-day study, whereas the patients with double concussion stated the same at the 30-day time point (although standardized symptom questionnaires were not administered and symptom assessments were not conducted at the intermediate time points). These findings were subsequently confirmed with a larger, multicenter cohort study of concussed athletes.
Additional markers of impaired mitochondrial reductive capacity include the lactate/pyruvate ratio, which is increased in patients with severe TBI. Therefore, it is logical to conclude that mitochondrial dysfunction increases the susceptibility for repeat injury. However, this has not been replicated in humans and MR spectroscopic data do not show statistically significant increases in the lactate ratios.
Axonal damage is observed following mild TBI, which has been observed clinically and proved experimentally. As mentioned earlier, white matter abnormalities have been described using DTI imaging after mild TBI in humans, although these findings are not universal. Similar to the metabolic changes, this injury is also worsened by repeat concussion TBI. This finding was shown by a repeat concussion animal model with a 3-day interval between injuries showing a significant increase in cytoskeletal damage and axonal injury. A more recent murine study assessing repetitive concussion (5 concussions, 48 hours apart) not only showed worsened motor and cognitive deficits but also noted axonal injury in the corpus callosum, which was worse in the repetitive injury arm. To date, there are no specific human studies of DTI conducted early after repeated concussive injuries.
Repeat mild TBI in animals acutely also induces spatial memory deficits (seen on MWM), and these impairments are related to impact severity and the number and timing of repeated injuries. An National Collegiate Athletic Association (NCAA) concussion study similarly showed that athletes who sustained repeat concussions (3 or more) were at a higher risk of an additional concussion. More importantly, these patients had a significantly longer duration of postconcussive symptoms than those athletes with only 1 concussion (30% vs 14.6%).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


