Essentials of Diagnosis
General Considerations
Pulmonary hypertension is among the most serious complications of rheumatic diseases. Pulmonary hypertension can be classified into one of five broad categories (Table 63–1). While rheumatic conditions can be associated with any classification of pulmonary hypertension (see Table 63–1), the most common category in which rheumatic conditions fall is pulmonary arterial hypertension—a pulmonary vasculopathy in the absence of left heart dysfunction, underlying parenchymal lung disease, thromboembolism, or other causes.
|
Connective tissue diseases accounted for one-quarter of all pulmonary arterial hypertension cases in REVEAL, a large US registry. Pulmonary arterial hypertension is particularly common in scleroderma or systemic sclerosis with a prevalence of 4–8%, and an estimated incidence of 0.61 cases per 100 patient years, accounting for 65% of connective tissue diseases associated with pulmonary arterial hypertension. In systemic sclerosis, pulmonary arterial hypertension typically occurs in older women (>80% female; mean age of 60 years) with limited scleroderma (up to 90%) and is more common among patients with late-onset systemic sclerosis. The average duration of systemic sclerosis prior to pulmonary arterial hypertension diagnosis varies from 4 to 14 years, but it can occur concomitant with or soon after disease onset.
The frequency of pulmonary arterial hypertension in other connective tissue diseases is not well characterized, but its occurrence is well recognized with lupus, representing 22% of all connective tissue disease–related pulmonary arterial hypertension, followed by rheumatoid arthritis at 9%. Features of systemic sclerosis are often present in such cases, ie, overlap syndrome or mixed connective tissue disease. In patients with lupus, the mean age at which pulmonary arterial hypertension is diagnosed is 46 years, and in the United States, most are nonwhite females.
Chronic interstitial lung disease is common in certain connective tissue diseases, particularly systemic sclerosis. Pulmonary hypertension complicates interstitial lung disease in about 30% of cases, depending on the extent of fibrosis. The combination of both pulmonary hypertension and interstitial lung disease portends a dramatically worse prognosis than either condition alone. In large series of systemic sclerosis patients undergoing evaluation for pulmonary hypertension, about one third of those with documented precapillary pulmonary hypertension had significant pulmonary fibrosis.
The most common type of pulmonary hypertension in the general population is that due to left heart disease, particularly nonsystolic left heart failure. Patients with rheumatic conditions may have left ventricular diastolic dysfunction associated with traditional cardiovascular risk factors or by virtue of direct cardiac involvement. Among patients with systemic sclerosis undergoing cardiac catheterization for suspected pulmonary arterial hypertension, 10–20% have elevated pulmonary artery wedge pressure indicative of left heart disease.
Chronic thromboembolic disease must be considered in any case of pulmonary hypertension, since surgical thromboendarterectomy can be curative. Antiphospholipid antibodies are present in 10% of such patients. Systemic vasculitides are rare causes of pulmonary hypertension, either due to direct vascular involvement or as a consequence of chronic parenchymal lung disease. In Takayasu arteritis, pulmonary hypertension has been noted in up to 12% of cases.
A high degree of suspicion for the diagnosis is required, since symptoms of dyspnea and fatigue are often nonspecific. A comprehensive clinical testing algorithm is required to confirm the diagnosis, exclude other causes, and assess disease severity. Recent advances in the therapy of pulmonary arterial hypertension have improved the outlook considerably, but mortality remains high. Collaboration with a pulmonary hypertension specialist is important to optimize long-term outcomes.
Pathology & Pathophysiology
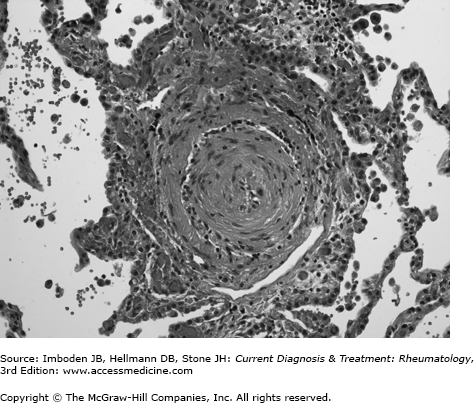
The pulmonary circulation is a low pressure, low resistance circuit designed to accommodate large changes in venous return with minimal increases in pressure. Over 50% of the vascular bed must be obstructed or destroyed before resting pulmonary artery pressure begins to rise. In pulmonary arterial hypertension, the site of obstruction is the small muscular pulmonary arteries and precapillary arterioles. Characteristic, although nonspecific, vascular lesions consist of concentric laminar and nonlaminar intimal fibrosis with lumenal obliteration (Figure 63–1) and eccentric intimal fibrosis, which may represent residua of thrombosis in situ. Medial hypertrophy and adventitial fibrosis may also be present to variable degrees. Plexiform lesions, which are complex glomerular-like sinusoidal channels lined by proliferating endothelial cells that may represent an attempt to bypass downstream obstruction or a precursor to concentric-obliterative lesions, are conspicuously rare or absent in systemic sclerosis-pulmonary arterial hypertension, compared with idiopathic pulmonary arterial hypertension. Another distinctive feature of systemic sclerosis-pulmonary arterial hypertension is the frequent occurrence of venous remodeling with features of pulmonary veno-occlusive disease. Focal perivascular accumulations of lymphocytes accompany the vascular lesions, but a true vasculitis is rare. With interstitial lung disease, vascular obliteration and destruction is routinely present in areas of fibrosis, with normal vessels elsewhere. When severe pulmonary hypertension is present, diffuse vascular remodeling is typically observed in the lung.
Pulmonary vascular obstruction induces abnormalities in both gas exchange and cardiac function. Reduced pulmonary blood flow relative to ventilation leads to wasted ventilation or dead space physiology. As a result, minute ventilation must rise to avoid hypercapnia. Indeed, one of the earliest and characteristic findings of pulmonary vascular disease on cardiopulmonary exercise testing is an increase in dead space fraction and minute ventilation relative to carbon dioxide production with exercise. Obliteration of the capillary bed limits transit time with resultant exercise-induced hypoxemia. Resting oxygen desaturation suggests right-to-left shunting (either intrapulmonary due to opening of arteriovenous communications or cardiac related due to opening of a patent foramen ovale as a result of raised right atrial pressure) or the presence of low ventilation/perfusion (V̇/Q̇) units. The latter occurs in the setting of parenchymal lung disease (eg, interstitial lung disease). Superimposed pulmonary vascular disease dramatically worsens hypoxemia associated with V̇/Q̇ mismatching because the ability of the lung to redirect blood flow to better ventilated units is impaired.
Increased pulmonary vascular resistance initially induces concentric right ventricular hypertrophy to compensate for the increased afterload. This allows the cardiac output to be normal at rest and even with exercise early in the disease course. With progressive increases in pulmonary vascular resistance, the right ventricle begins to dilate, inducing a volume load due to tricuspid regurgitation, in addition to the pressure load. Stroke volume and ultimately cardiac output is unable to increase appropriately with exercise and begins to fall at rest. Right atrial pressure rises, leading to systemic vascular congestion. Massive dilatation of the right ventricle compromises left ventricular filling, further impairing cardiac performance. The dilated, hypertensive right ventricle has an increased oxygen demand. This, combined with low systemic blood pressure (resulting in reduced right coronary perfusion pressure gradient), creates a relative right ventricular ischemia that likely plays a role in the development of progressive right heart failure, the primary cause of death in pulmonary arterial hypertension.
Clinical Findings
Dyspnea with activity is the most common complaint in pulmonary arterial hypertension and is the presenting symptom in most cases. The degree of functional limitation based on the New York Heart Association classification has important prognostic implications. Fatigue, chest pain, exertional syncope or near-syncope, palpitations, and leg edema are other frequent complaints. In systemic sclerosis, skin thickening may reduce the tendency for leg edema, and fluid may preferentially accumulate in the abdomen.
Pulmonary hypertension complicating interstitial lung disease is often heralded by abrupt worsening of dyspnea that had been previously stable for years and by an increase or new requirement for supplemental oxygen. Diffuse cutaneous involvement is present in over half of patients who have combined interstitial lung disease and pulmonary hypertension. Raynaud phenomenon and antiphospholipid antibodies (in the absence of thromboembolism) are more prevalent in lupus associated–pulmonary arterial hypertension compared with lupus patients without pulmonary arterial hypertension.
Physical examination findings of pulmonary hypertension include a left parasternal lift of right ventricular hypertrophy, an accentuated pulmonic component of the second heart sound, a holosystolic murmur of tricuspid insufficiency and a right ventricular gallop. Elevated jugular venous pressure, hepatomegaly, ascites, and lower extremity edema indicate right ventricular decompensation. Systemic hypertension suggests left ventricular diastolic dysfunction as the potential cause of pulmonary hypertension, whereas low blood pressure may signify right heart failure. Bibasilar crackles are often present with interstitial lung disease.
A structured diagnostic approach is required to establish the correct diagnosis. Patients with symptoms and signs suggestive of pulmonary hypertension should undergo initial evaluation with a chest radiograph, pulmonary function testing, and Doppler echocardiography. The latter is a useful screening tool for pulmonary hypertension. It can demonstrate right heart enlargement (Figure 63–2) and right ventricular hypokinesis as well as provide an estimate of right ventricular (and hence pulmonary artery) systolic pressure (RVSP) based on the modified Bernoulli equation where RVSP = 4v2 + right atrial pressure estimate, where v is the maximal tricuspid regurgitant velocity. The upper limit of normal for RVSP is 35 mm Hg and increases with age. However, RVSP estimates can be inaccurate, with both overestimations and underestimations of more than 10–20 mm Hg common. Another potential source of error is the right atrial pressure estimate. Many echocardiogram laboratories routinely add 10 mm Hg for right atrial pressure, whereas the normal right atrial pressure in the absence of right heart dysfunction is 3–5 mm Hg. As a result, the sensitivity and specificity of echocardiography using a cutoff RVSP of 40 mm Hg for detecting pulmonary hypertension confirmed by invasive hemodynamics can be as low as 75% and 60%, respectively. Thus, echocardiographic findings should not be solely relied upon to diagnose or exclude pulmonary hypertension but rather should be combined with other clinical features to ascertain the likelihood of pulmonary hypertension. Other echocardiographic findings suggestive of pulmonary hypertension include flattening of the interventricular septum and reduced pulmonary artery acceleration time. Doppler echocardiography also provides important information on left heart structure and function as a potential cause for pulmonary hypertension. Pericardial effusion is present in roughly one third of patients with pulmonary arterial hypertension-systemic sclerosis and is associated with increased mortality, likely reflecting increased right atrial pressure and right ventricular failure.
Figure 63–2.




