Chapter 200 Porphyrias
 Diagnostic Summary
Diagnostic Summary
• Unexplained abdominal pain, possibly including nausea and vomiting
• Acute peripheral or central nervous system dysfunction
• Mental abnormalities ranging from confusion to acute psychosis
• Urinary porphobilinogen during attack
• Most carriers are usually asymptomatic except when exposed to drugs or chemicals that exacerbate the condition
 General Considerations
General Considerations
Porphyria as a disease was first described three centuries ago by Sir Theodore Turquet de Mayerne of France, physician to the English court of King James I. He described accounts of paralysis, bilious urine, colic, insanity fits, permanent insanity, deaths at childbirth, and premature death in those afflicted. The disease probably afflicted King George III of England and was thought to have impaired his judgment during the American Revolution.1
In 1911, Günther described what is now known as the classic triad of abdominal pain, constipation (or diarrhea), and vomiting. Waldenström later added to the understanding of these diseases by describing acute intermittent porphyria (AIP) as characterized by periods of exacerbation and remission.2
The genetic deficits of porphyria affect all tissues but primarily the blood-forming tissues of the bone marrow and the cytochrome P-450 system in the liver because of the greater number of porphyrin precursors required. The liver is the major site where inherited or acquired deficits in heme synthesis occur.3
Classifications of Porphyria
Acute hepatic porphyrias are characterized by a rapid onset of symptoms that are largely neurologic; the erythropoietic variety manifests primarily in the skin, leading to cutaneous photosensitivity (Box 200-1).
Signs and Symptoms of Hepatic Porphyrias
Acute attacks are comprised of a wide variety of symptoms, ranging from skin lesions to abdominal pain to neurologic manifestations of varying intensity. Abdominal, back, or extremity pain grows in intensity over a 24- to 48-hour period, which may lead the clinician to consider appendicitis or an acute abdomen. Rebound tenderness is not usually present.
Mental abnormalities can range from confusion to acute psychosis and may be the first presentations of an attack. With prolonged attacks, sensory and motor functions are impaired and can result in respiratory paralysis and death.5 Many patients are diagnosed with schizophrenia, and a considerable number confined to mental institutions are thought to be afflicted with one of the porphyrias.6,7
 Biochemistry of Porphyria
Biochemistry of Porphyria
Heme Formation
Red blood cell (RBC) incorporation of heme, iron, and glycine occurs in maturing cells up through and including reticulocytes but is eventually lost as the red cell ages. Hypoxia and erythropoietin will increase ALA synthase activity in RBCs but not the liver, whereas drugs and chemicals will affect the liver but not erythropoietic tissues. Figure 200-1 illustrates heme synthesis and porphyria.
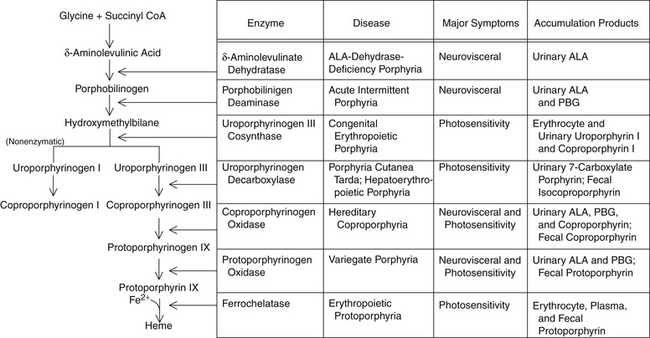
 Manifestation of Porphyria
Manifestation of Porphyria
Cutaneous manifestations occur because some porphyritic precursors absorb light at 400 nm, resulting in photosensitivity. The absorption of light raises the potential energy of the molecule to an “excited” state, resulting in a highly reactive oxygen species. The release of histamine and proteolytic enzymes then results in oxidative damage. Beta-carotene protects against these injuries.3,4,8
Neuropsychiatric changes occur in the hepatic porphyrias (ALA-dehydrase-deficiency porphyria, AIP, porphyria cutanea tarda, hereditary coproporphyria, and variegate porphyria) with excess production of ALA and porphobilinogen (PBG), and there is a somewhat linear relationship between their concentration and the severity and duration of symptoms. A buildup of porphyrin intermediaries has been found in various tissues and may induce the neuropsychiatric symptoms accompanying acute flareups.9,10 The exact mechanism whereby porphyrins affect the central nervous system is not well understood.
Patients with familial porphyria cutanea tarda (PCT) usually present at a younger age than those who develop PCT spontaneously, despite the fact that there is no difference in the biochemical features of the disease.11 This may be because of a corresponding genetic predisposition to sequester ferritin in the liver or an increased sensitivity to ethanol ingestion.11,12 Additionally, even in families with known genetic deficits, manifestations of the disease among family members vary.
Etiologic Agents Triggering Porphyria
Numerous agents that trigger episodes of porphyria have been identified, and patients afflicted must be careful when taking prescription medications, estrogens, and some herbal medicines; they must also avoid environmental exposures to heavy metals, organophosphates, and any substance that places an excess burden on the cytochrome P-450 system. In particular, exogenous estrogens—whether from oral contraceptives, estrogen patches, or estrogen replacement therapy—are strong contributors to an underlying porphyria deficit.13,14 This is one reason why acute attacks of porphyria are seen more often in women than men, especially premenstrually. Additionally, some studies have suggested that estrogens enhance the porphyrin-inducing activities of other agents, making women more vulnerable to environmental exposures than their male counterparts.15,16
Herbicide-induced porphyria has been shown to decrease the activity of several enzymes involved in the porphyritic pathway as well as to increase the porphyrin content in nerve tissue.16,17 A considerable number of chemicals have also been linked to porphyria or porphyrinuria in humans; these generally involve chronic industrial exposures or environmental exposures. An example of an epidemic of PCT produced by the accidental ingestion of wheat treated with the fungicide hexachlorobenzene occurred in Turkey in the 1950s.
Some researchers have hypothesized that several otherwise unexplained chemical-associated illnesses such as multiple chemical sensitivity syndrome (MCSS) may represent mild chronic cases of porphyria or other acquired abnormalities in heme synthesis.18,19
According to William Morton, who has written extensively on porphyrias, MCSS as first described by Cullen in 1979 may, in fact, be porphyria. Morton speculates that exposures to porphyrogenic chemicals, medications, or severe infection overpower the already deficient enzyme system, resulting in an accumulation of the specific porphyrin because of diminished enzyme function. He proposed that the resultant symptoms are due to an increase of the porphyrinogen and not an accumulation of the toxin itself. Many diagnosticians have tried to expedite the distinction between “real” porphyria and secondary porphyrinopathies by requiring that a porphyria diagnosis be based on urinary or fecal porphyrin excretions or both 2 to 20 times the upper limit of normal. This classification would exclude individuals demonstrating lesser degrees of porphyrinogenic activity, possibly some of those whose conditions are in remission or not subject to environmental exposures.20
The intensity of porphyria symptoms varies widely. About 10% of the cases present with acute severe attacks, whereas approximately 25% are seen with chronic symptomatology of varying degrees. The remaining percentage (65%) present with no symptoms but may become susceptible under the right circumstances.21
Not all researchers agree with Morton’s and others’ premise that environmental toxicities induce underlying genetic deficits leading to symptomatic porphyria. According to Hahn and Bonkovsky, “patients with multiple chemical sensitivity syndrome may, at times, have modest increases in urinary coproporphyrin excretion; this is a common finding found in many asymptomatic subjects or patients with diverse other conditions (e.g., diabetes mellitus, heavy alcohol use, liver disease, and many kinds of anemia). Such secondary coproporphyrinuria does not indicate the existence of coproporphyria.”22
Regardless of which view is correct, an increasing number of cases are being diagnosed by clinicians. Certainly, with the increasing levels of pollutants in the environment, hormonal additives to the food chain, and the unmonitored multiple pharmacy prescriptions encountered by most humans, the potential for unmasking an underlying deficit increases.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


