CHAPTER 12 Pharmacology of Local Anesthetic Agents
INTRODUCTION
A major early influence on the development of cocaine as a local anesthetic was a paper published in 1880 by Vassiliy von Anrep, in which he described the pharmacology of cocaine and suggested its use for surgery. Anrep systematically investigated its effects on different tissues in frogs and various mammals. He described the stimulating effect when cocaine was administered systemically and the depressant effect at high doses, with death apparently from respiratory arrest. Although Moréno and Bennett had previously investigated the effects of cocaine in animals, and Moréno had described the blocking action of cocaine when injected near the sciatic nerve in frogs and suggested its use as a local anesthetic, Anrep’s study was much more detailed and comprehensive, going through all the body’s systems methodically in different species. He was the first to inject cocaine subcutaneously into humans (himself), and the first to report the anesthetic effect produced by the injection. (He described the feeling of warmth and numbness which persisted for 25–30 minutes.) He also confirmed the numbing effect when cocaine was placed on the tongue. Anrep suggested that these properties could have very important applications in surgery and the treatment of painful conditions, concluding that thus far he had been too busy to explore them.1
The news spread quickly and only 2 weeks later, Jellinek reported similar anesthetic and analgesic effects when cocaine solution was applied to the mucous membrane of the pharynx and larynx. Over the next 2 months many other investigators from all over the world published reports of the use of topical cocaine for anesthetizing the ears, nose, mouth, trachea, rectum, and genital tract. On November 15, Hepburn published his observations on the numbing effect of cocaine after hypodermic injection. Other reports followed throughout November, 1884. On December 6, 1884, Hall announced his and Halsted’s experiences in producing nerve blockade with cocaine (ulnar nerve, musculocutaneous nerve of the leg, anterosuperior dental nerve, inferior dental nerve, and lingual nerve) in a letter dated November 26. Many reports of cocaine’s use followed, mostly involving topical application and/or infiltration.1
Local and regional anesthesia were born!
The introduction of cocaine into medical practice by Koller revolutionized surgery, starting with ophthalmology and spreading rapidly to urology, gynecology, otolaryngology, and general surgery, as well as to dentistry. The use of local and regional anesthesia remained uncommon for a few more years until the synthesis of pure cocaine in 1891 by the techniques of modern organic chemistry.2 Soon, however, the toxic effects of cocaine, resulting in the deaths of many patients as well as addictions among the medical staff, were also identified. The need for better and safer local anesthetics became apparent.
Procaine, an alternative to cocaine, belonging to the class of structures called amino esters, appeared in 1905. Other amino esters synthesized between 1891 and 1930 included tropocaine, eucaine, holocaine, orthoform, benzocaine, and tetracaine. Marketed under the name Novocaine™, procaine, which demonstrated predictable effects and low systemic toxicity, remained the most important local anesthetic until the 1940s.. However, the relatively frequent allergic reactions to Novocaine™, due to the metabolite p-aminobenzoic acid (PABA), prompted the search for newer, less allergenic substances.
In 1908, intravenous regional anesthesia was first described by August Bier. This technique is still in use and is remarkably safe when drugs of relatively low systemic toxicity such as lidocaine and prilocaine are used. Spinal anesthesia was first described in 1885 but not introduced into clinical practice until 1899, when August Bier subjected his assistant and himself to a clinical experiment in which they observed the anesthetic effect, but also the typical side effect, of postpunctural headache. Within a few years, spinal anesthesia became widely used for surgical anesthesia and was accepted as a safe and effective technique. Although atraumatic (non-cutting-tip) needles and modern drugs are used today, the technique has otherwise changed very little over many decades.3
A new substance, lidocaine, was first developed in 1943 and marketed in 1947 under the name Xylocaine™. It was the first amino amide local anesthetic, and it carried a significantly reduced risk of allergic reactions. Other amide local anesthetics including nirvaquine, cinchocaine, mepivacaine, prilocaine, efocaine, bupivacaine, etidocaine, and articaine were ostensibly less toxic than cocaine, but they had differing amounts of central nervous system (CNS) and cardiovascular (CV) toxicity. Bupivacaine is of special interest because of its high potency, long duration of action, and history of clinical application. It was synthesized in 1957 and first marketed in 1965. However, increasing reports of CNS and CV toxicity, and especially the identification of special therapy-resistant cardiotoxicity, led to restriction of its use. The numerous experimental studies that followed to identify the cellular mechanism of this toxicity increased our understanding of the action of local anesthetics greatly.2
Thereafter, further impetus to the synthesis and clinical introduction of other amide-type local anesthetics came from the continuing development of regional anesthesia and pain medicine. The identification of optically active isomers of the mepivacaine family and technological advances led to the ability to selectively produce the pure S-(-) enantiomer of ropivacaine, whose toxicology was extensively studied before its introduction on the market in 1996. During extensive clinical use unwanted side effects have been limited.2 Ropivacaine is structurally related to bupivacaine and mepivacaine. However, it possesses a different pharmacodynamic profile, specifically on cardiac electrophysiology (less arrhythmogenic than bupivacaine). Studies on the anesthetic activities and toxicity of the individual enantiomers of bupivacaine and mepivacaine generally indicate that the S-enantiomers are less toxic than the R-enantiomers.4
MECHANISM OF ACTION
Pharmacokinetics and pharmacodynamics
Local anesthetics are frequently applied directly on or near their target via techniques of regional anesthesia. Thus, plasma levels rarely bear a relationship to their desired effects; however, these plasma levels are critical for systemic toxicity. The pharmacokinetics describing the blood concentration after administration of a drug are illustrated in Figure 12.1. Clearance of amide-type local anesthetics depends on liver function and may be limited in any disease state that reduces hepatic blood flow. An increase of α-1-acid glycoprotein and other plasma proteins (e.g. due to cancer) can lead to more rapid delivery of the drug to the liver and therefore lead to faster clearance. Ester-type drugs are hydrolyzed by plasma pseudocholinesterase. Thus, patients with genetically abnormal pseudocholinesterase are at increased risk for toxic side effects, since metabolism is slower. However, to a limited degree, the liver also extracts and metabolizes ester-type compounds. An increase of apparent volume of distribution may occur in older and more adipose patients. The lung has also been implicated as a site of local anesthetic metabolism, providing a dose-dependent first-pass uptake effect. Renal disease has little effect on these pharmacokinetics.
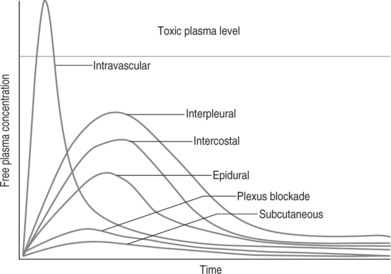
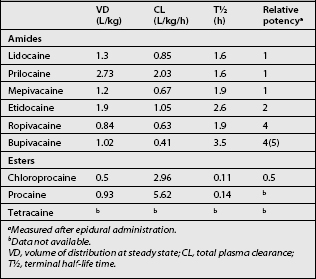
Table 12.1 summarizes some pharmacokinetic and -dynamic parameters for amide- and ester-type local anesthetics.
Transport to the ion channel
Most local anesthetics are weak bases that are converted to their uncharged lipid-soluble form to pass through the phospholipid membrane into the cell as shown in Figure 12.2. However, once inside they are converted to the charged form to block the sodium channel. The Henderson-Hasselbalch equation relates the pH to the pKa and the relative concentrations of uncharged base (B) and charged base (BH+). Given the reaction:
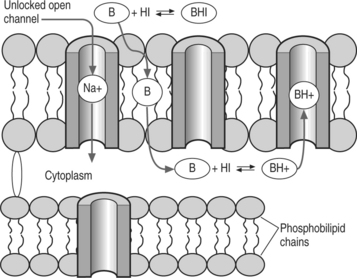
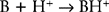
The pH determines the ratio of B and BH+:
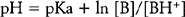
By definition, the pKa is the pH at which 50% of the local anesthetic is uncharged ([B]=[BH+]). As shown in Figure 12.3, the pKa range for commonly used local anesthetics lies in a narrow band.
Modulation of pH by carbonation
The addition of bicarbonate has also been used to increase the pH of local anesthetic solutions, thus increasing the concentration of nonionized lipophilic free base. This will theoretically increase the rate of diffusion of the drug and its speed of onset. Figure 12.4 illustrates this concept using lidocaine. Clinically, the addition of 1 mEq of sodium bicarbonate to each 10 mL of commercially prepared 1.5% lidocaine solution produces a significantly faster onset of anesthesia and more rapid spread of sensory block.5

Table 12.2 lists several common local anesthetics with their pKa values and their usual pH in storage.
Table 12.2 pKa and pH of commonly used local anesthetics
| Drug | pKa | pH of solution in vial |
|---|---|---|
| 2-chloroprocaine | 9.1 | 2.5–4 |
| Procaine | 8.9 | 5.5–6 |
| Tetracaine | 8.4 | 4.5–6.5 |
| Cocaine | 8.5 | variable |
| Lidocaine | 7.9 | 5.6 (without epi) |
| 3.5–5.5 (with epi) | ||
| Levobupivacaine | 8.1 | 4.0–6.5 (without epi) |
| Bupivacaine | 8.1 | 4.5–5.5 (without epi) |
| 3.5–5.5 (with epi) | ||
| Mepivacaine | 7.7 | 5.5 |
epi, epinephrine.
Molecular mechanism of sodium channel blockade
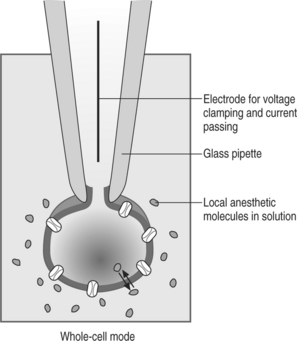
The generation and propagation of impulses in nerve axons to carry afferent (sensory) and efferent (motor, sympathetic) information requires the flow of specific ionic currents through channels in the plasma membrane. These channels open and close depending on the electrical potential of the cell membrane. The major determinant of the depolarization of nerve fibers is the specific influx of Na+ ions through sodium channels located in nerve axons. Local anesthetic agents reversibly bind to and block sodium channels, thereby preventing the initiation or propagation of the electrical impulses required for nerve conduction. It now appears that although the uncharged form of a local anesthetic is more likely to cross into the cell membrane as mentioned above, either form may bind to the Na+ channel. The local anesthetic blocks the channel by binding to the receptor from inside the cell. A well-established technique in electrophysiology, the patch-clamp technique (Figure 12.5, illustrating the ‘whole-cell’ mode of this technique), was instrumental in explaining the basic molecular mechanism of Na+ channel blockade caused by LAs. Most LA enter the channel from cell cytoplasm when the channel has been activated to the ‘open’ state. Upon blocking the flow of sodium ions, the channel enters the ‘inactivated’ state. LA bind tightly to and stabilize the inactivated state.
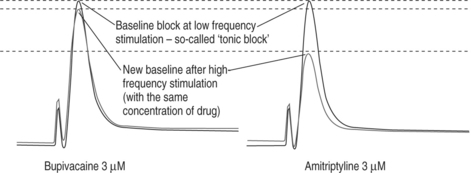
Use-dependent block (additional block when stimulating at a high frequency)
Local anesthetics such as lidocaine and bupivacaine exhibit a phenomenon called use dependency.6,7 This means that if a neuronal cell is stimulated at a low frequency of 0.03 Hz (e.g., once every 30 sec) at a given concentration, a certain amount of inhibition of Na+ current will ensue and establish a steady state block. This block is called a tonic block and is thought to be equivalent to the regional/local block performed in the operating room when there is no high-frequency stimulation by pain fibers (before surgery). If, however, under the same conditions (same concentration of drug) the frequency is increased (e.g., 5 Hz, or once every 200 msec), an additional block will occur, and a new steady state will result. This interesting feature is called use-dependent block. It is seen with all local anesthetics, and is particularly prominent with amitriptyline, which also has local anesthetic properties, and has been commonly used orally for neuropathic pain. However, due to a very narrow therapeutic ratio, amitriptyline has no clinical use as a local anesthetic. It is thought that bupivacaine and amitriptyline have a higher affinity to the LA binding site than, for example, lidocaine, and therefore at higher stimulation frequencies there is not enough time for the drug to dissociate from the binding site and leave the channel.8 The concept of use-dependent block is illustrated in Figure 12.6, depicting amitriptyline versus bupivacaine, as the former also has significant local anesthetic properties.
Clinical relevance of use-dependent block
With the currently available, and also some of the experimental local anesthetics, relatively little tonic block can be found at low concentrations.9 This suggests that discharge at a high rate from stimulated sensory/pain fibers could be blocked, while leaving other nerve fibers (motor) undisturbed. Therefore, this property may make it possible to selectively block the ‘firing’ of high-frequency action potentials produced by pain fibers while leaving the ‘normal’ action potential transmission intact.
For the same reason, drugs inducing a high degree of use-dependent block, when given at higher concentrations, should prolong the duration of a complete block. In other words, a drug with a high degree of use-dependent blockade could give a differential block, i.e. a selective block of specific (pain-transmitting) nerve fiber groups10 at lower concentrations, and a prolonged complete block at higher concentrations (similar to bupivacaine, but to a much greater extent). Preclinical in vivo11 and clinical studies12 appear to confirm the role of use-dependent block in anesthesia/analgesia.
EFFECTS OF LOCAL ANESTHETICS OTHER THAN NA+ CHANNEL BLOCKADE
Local anesthetics also have been shown to inhibit K+ channels, Ca2+channels, nicotinic acetylcholine-activated conductance, the substance P receptor, and even the G protein modulation of certain channels. These alternative actions may contribute to local anesthesia and to some aspects of toxicity.
Inhibition of signaling through G protein-coupled receptors
Local anesthetics have may have significant effects in several settings other than local and regional anesthesia or antiarrhythmia. Some of these effects occur at concentrations that are much lower than those required for Na+ channel blockade. For example, the half-maximal inhibitory concentration (IC50) of lidocaine at the neuronal Na+ channel is approximately 50–100 mM (depending on the specific channel subtype and study preparation), whereas the compound inhibits signaling through M1 muscarinic receptors (expressed recombinantly in Xenopus laevis oocytes) with an IC50 of 20 nM, that is, 1000- to 5000- fold lower. This greatly increased sensitivity of other targets implies that at relatively low LA concentrations (such as attained in blood during epidural anesthesia or analgesia or during intravenous LA infusion) other significant pharmacologic effects may be present.13
Interactions of LAs with neutrophil signaling
Polymorphonucleocytes (PMNs) do not express Na+channels, and LA effects on these cells therefore are not caused by Na+channel blockade. LA effects on these cells are also not affected by experimental Na+ channel blockers such as tetrodotoxin or veratridine. Overactive inflammatory responses that destroy rather than protect are critical in the development of a number of perioperative disease states, such as postoperative pain, adult respiratory distress syndrome (ARDS), systemic inflammatory response syndrome, and multiorgan failure. Perioperative modulation of such responses is therefore relevant to the practice of anesthesiology and interventional spine medicine, and LAs may play significant roles in this regard.13
Leukotriene B4, formed in inflammatory cells such as PMNs and monocytes, is a potent stimulator of PMN activity and therefore has an important role in inflammation.14 It induces margination at endothelial cells, degranulation, diapedesis, and superoxide generation and acts synergistically with prostaglandin E2 to enhance vascular permeability. Therefore, renewed interest has been created following the discovery that LAs block leukotriene release.15
Similarly, interleukin (IL)-1α is another inflammatory mediator, which acts on its receptor on PMNs, stimulating phagocytosis, respiratory burst, chemotaxis, and degranulation. Lidocaine and bupivacaine dose-dependently inhibit IL-1α release in lipopolysaccharide-stimulated human peripheral blood mononuclear cells.15
Effects of local anesthetics on polymorphonucleocyte adhesion
Excessive adhesion of PMNs to endothelium may induce endothelial injury, which is mediated by several adhesion molecules. One of the most important for firm adhesion of PMN to endothelium and subsequent transmigration (diapedesis) is CD11b–CD18, a member of the integrin family. This receptor is expressed constitutively on the surface of nonactivated PMN, but expression increases markedly after inflammatory stimulation. Binding of activated PMN to endothelial cells by CD11b–CD18 increases intracellular peroxide levels in the endothelial cells, in which reactive oxygen species can have detrimental effects. Monoclonal antibodies against CD11b–CD18 protect in vitro against endothelial cell injury. In vitro studies have shown a reduction of TNF-α-induced upregulation of CD11b–CD18 surface expression on PMN after ropivacaine or lidocaine treatment. This may contribute to the beneficial in vivo effects of ropivacaine on ulcerative colitis at tissue concentrations (100–300 mM) obtained after rectal LA administration.13
Effects of local anesthetics on PMN priming
It is possible that inhibition of priming contributes to the antiinflammatory actions of LAs, and in particular suppresses the deleterious effects of the uncontrolled, overactive response of inflammatory cells to a stimulating agent. This might explain how LAs can decrease tissue damage without significantly inhibiting PMN functions required for host defense. In summary, LAs do not scavenge the reactive oxygen species generated, but rather inhibit the ability of PMNs to produce them.13
SUMMARY OF LOCAL ANESTHETIC AGENTS
General considerations
Clinically used local anesthetics may be classified as amino esters (procaine, chloroprocaine, tetracaine, and cocaine) or amino amides (lidocaine, mepivacaine, prilocaine, bupivacaine, ropivacaine, levobupivacaine) as shown in Table 12.1 (Fig. 12.7).
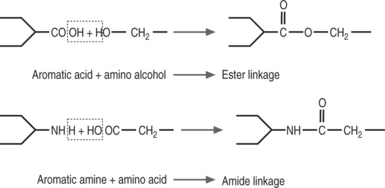
Ester and amide local anesthetics differ in their chemical stability, metabolism, and allergic potential. Amides are extremely stable, whereas esters are relatively unstable in solution. Esters are hydrolyzed in plasma by the cholinesterase enzymes, whereas the amide compounds undergo enzymatic degradation in the liver. Cocaine is an exception to this as it is metabolized predominantly by the liver. The metabolites of esters include p-aminobenzoic acid (PABA), which can rarely induce allergic-type reactions. Allergies to amides are extremely rare. Tables 12.3–12.8 list usual doses of local anesthetics based on the type of block. A brief synopsis of commonly used local anesthetics follows below.
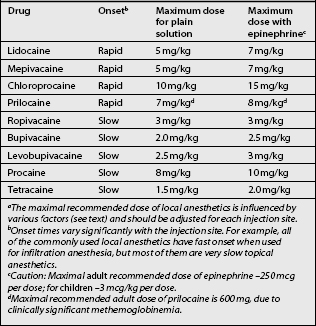
Table 12.4 Tissue infiltration
| Drug | Volume for 70 kg person (mL) | Usual duration (min) |
|---|---|---|
| Chloroprocaine 1–2% plain | 70–35 | 15–30 |
| Chloroprocaine 1–2% with epinephrinea (1:200 000–1:400 000) | 105–53 | 30–60 |
| Prilocaine 0.5–1% plain | 100–50 | 30–90 |
| Prilocaine 0.5–1% with epinephrinea (1:200 000–1:400 000) | 112–56 | 60–120 |
| Lidocaine 0.5–1% plain | 70–35 | 30–60 |
| Lidocaine 0.5–1% with epinephrinea (1:200 000–1:400 000) | 100–50 | 60–120 |
| Mepivacaine 0.5–1% plain | 70–35 | 45–90 |
| Mepivacaine 0.5–1% with epinephrinea (1:200 000–1:400 000) | 100–50 | 60–120 |
| Bupivacaine 0.25–0.5% plain | 56–28 | 120–180 |
| Bupivacaine 0.25–0.5% with epinephrinea (1:200 000–1:400 000) | 70–35 | 150–240 |
| Levobupivacaine 0.25–0.5% | 70–35 | 120–180 |
| Levobupivacaine 0.25–0.5% with epinephrinea (1:200 000–1:400 000) | 84–42 | 150–240 |
| Ropivacaine 0.2–0.5% | 105–42 | 90–150 |
Note: do not exceed maximum dosage listed in Table 12.3.
a Caution: Maximal adult recommended dose of epinephrine –250 mcg per dose; for children –3 mcg/kg per dose.
Table 12.5A Minor nerve blocks (single nerve such as ulnar or radial, blocks below the knee)
| Drug | Volume (mL) | Usual duration (min) |
|---|---|---|
| Lidocaine 1–2% plain | 5–10 | 60–120 |
| Lidocaine 1–2% with epinephrinea (1:200 000–1:400 000) | 5–10 | 120–180 |
| Chloroprocaine 2–3% plain | 5–10 | 15–30 |
| Chloroprocaine 2–3% with epinephrinea (1:200 000–1:400 000) | 5–10 | 30–60 |
| Prilocaine 1–2% plain | 5–10 | 60–120 |
| Prilocaine 1–2% with epinephrinea (1:200 000–1:400 000) | 5–10 | 120–180 |
| Mepivacaine 1–1.5% plain | 5–10 | 60–120 |
| Mepivacaine 1–1.5% with epinephrinea (1:200 000–1:400 000) | 5–10 | 120–360 |
| Bupivacaine 0.25–0.5% plain | 5–10 | 180–480 |
| Bupivacaine 0.25–0.5% with epinephrinea (1:200 000–1:400 000) | 5–10 | 240–960 |
| Levobupivacaine 0.25–0.5% plain | 5–10 | 180–480 |
| Levobupivacaine 0.25–0.5% with epinephrinea (1:200 000–1:400 000) | 5–10 | 240–960 |
| Ropivacaine 0.2–0.5% | 5–10 | 120–480 |
Note: most minor nerve blocks are performed without the addition of epinephrine. Epinephrine is contraindicated in blocks of the digits or penis because tissue ischemia may result. Also, do not exceed maximum dosage listed when blocking several nerves.
a Caution: Maximal adult recommended dose of epinephrine −250 mcg per dose; for children − 3 mcg/kg per dose.
Table 12.5B Major nerve blocks (several nerves or a plexus)
| Drug | Volume (mL) | Usual duration (min) |
|---|---|---|
| Lidocaine 1–2% plain | 50–30 | 60–180 |
| Lidocaine 1–2% with epinephrinea (1:200 000–1:400 000) | 50–30 | 120–240 |
| Prilocaine 1–2% plain | 50–30 | 60–180 |
| Prilocaine 1–2% with epinephrinea (1:200 000–1:400 000) | 50–30 | 120–300 |
| Mepivacaine 1–1.5% plain | 50–30 | 60–180 |
| Mepivacaine 1–1.5% with epinephrinea (1:200 000–1:400 000) | 50–30 | 180–300 |
| Bupivacaine 0.25–0.5% plain | 40–20 | 180–720 |
| Bupivacaine 0.25–0.5% with epinephrinea (1:200 000–1:400 000) | 50–30 | 360–1440 |
| Levobupivacaine 0.25–0.5% plain | 50–30 | Same numbers as for bupivacaine |
| Levobupivacaine 0.25–0.5% with epinephrinea (1:200 000–1:400 000) | 50–30 | |
| Ropivacaine 0.5–0.75% plain or with epinephrinea (1:200 000–1:400 000) | 50–30 | 360–720 |
Note: do not exceed maximum dosage listed in Table 12.3.
a Caution: Maximal adult recommended dose of epinephrine − 250 mcg per dose; for children − 3 mcg/kg per dose.
Table 12.6 Epidural anesthesia
| Drug | Volume (mL) | Usual duration (min) |
|---|---|---|
| Chloroprocaine 2–3% plain or with epinephrine (1:200 000) | 30–15 | 30–60 |
| Lidocaine 1–2% plain | 25–15 | 45–60 |
| Lidocaine 1–2% with epinephrine (1:200 000) | 25–15 | 60–75 |
| Prilocaine 1–3% plain | 20–10 | 45–60 |
| Prilocaine 1–3% with epinephrine (1:200 000) | 20–10 | 60–180 |
| Mepivacaine 1–2% plain | 25–15 | 45–75 |
| Mepivacaine 1–2% with epinephrine (1:200 000) | 25–15 | 60–180 |
| Bupivacaine 0.25–0.75% plain | 20–10 | 90–240 |
| Bupivacaine 0.25–0.75% with epinephrine (1:200 000) | 25–10 | 120–300 |
| Levobupivacaine 0.25–0.75% plain | 25–10 | Same numbers as for bupivacaine |
| Levobupivacaine 0.25–0.75% with epinephrine (1:200 000) | 30–10 | |
| Ropivacaine 0.2–1.0% plain or with epinephrine (1:200 000) | 20–10 | 90–300 |
Note: do not exceed the maximum recommended dose listed in Table 12.3. Administration in fractionated doses of 3–5 mL is recommended.
Table 12.7 Spinal anesthesia – commonly used agents/dosesa
| Drug | Dose (mg)b | Usual durationc (min) |
|---|---|---|
| Procaine 5–10% | 80–160 | 30–75 |
| Lidocaine 1–2% | 20–100 | 30–90 |
| Mepivacaine 1.5–4% | 20–80 | 30–90 |
| Bupivacaine 0.5–0.75% | 5–18 | 75–300 |
| Levobupivacaine: same as bupivacaine | ||
| Ropivacaine 0.5–1% | 8–18 | 75–300 |
a While the injected volume of local anesthetic solution (with sufficiently high concentration) is critical in providing successful epidural, plexus, and peripheral nerve blockade, it is the actual local anesthetic dose (mg) that defines effective spinal anesthesia.
b The choice of appropriate local anesthetic dose for spinal anesthesia is influenced by the desired block level (segmental spread) and duration.
c Duration (and spread) is also dependent on baricity of spinally injected local anesthetic solution.
Table 12.8 Miscellaneous blocks
| Block | Drug | Volume/dose |
|---|---|---|
| Epidural roots | Bupivacaine 0.25% | 1–1.5 mL |
| Facet joint blockade | Bupivacaine 0.25% | 2–2.5 mL |
| Intra-articular facet joint injection | Bupivacaine 0.25% | Less than 1 mL as the joint capacity is limited |
| Epidural lidocaine and steroid injection | Prednisolone and lidocaine | 3 mL of lidocaine 0.5% with 3 mL of methylprednisolone (40 mg/mL) |
| Peripheral nerve, e.g. occipital nerve | Bupivacaine 0.25–0.5% | 2–5 mL |
| Trigger point | Bupivacaine 0.25–0.5% | 1–2 mL |
| Intravenous regional anesthesia | 0.5% lidocaine | 30–40 mL |
| Intravenous lidocaine infusion | 5 mg/kg over 45 min | |
| Intrathecal therapy | Lidocaine 2% without preservatives | Start 3 mg/day, increase by 20% per week till analgesia |
| Intra-articular facet joint injection | Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|





