Osteosarcoma
To qualify as an osteosarcoma, a neoplasm should have proliferating malignant cells that produce either osteoid substance or material histologically indistinguishable from it at least in small foci. Although the production of osteoid matrix is implicit in a diagnosis of osteosarcoma, this production may be quite focal and, hence, may not be recognized in limited tissue sampling. Thus, the diagnosis of osteosarcoma may be reasonable, even when no definite osteoid matrix is recognized if the neoplasm in question has features that in all respects are classic for osteosarcoma. In a qualifying tumor that is sampled throughout, elements with osteoid, chondroid, or fibromatoid differentiation may be predominant. Accordingly, in this series, osteosarcomas are divided into osteoblastic, chondroblastic, and fibroblastic types, depending on the dominant element. The implication is that what appears to be a chondroblastic osteosarcoma on biopsy may turn out to be an osteoblastic osteosarcoma when more tissue is sampled. This classification merely highlights the wide variation seen in the histopathology of osteosarcoma. It almost surely has no prognostic significance. All these tumors, however, are similar in the characteristics of bones of predilection, age of affected patients, pronounced tendency to early hematogenous dissemination, and necessity for prompt ablative surgical therapy. Malignant fibroblastic tumors with no definite osteoid production by neoplastic cells, regardless of their degree of anaplasia, are classified as fibrosarcomas or malignant fibrous histiocytomas. Similarly, chondroblastic malignant tumors with no definite sheets of spindle cells or osteoid production are designated chondrosarcomas. Sometimes exact designation is difficult and must be arbitrary because there is no special stain for osteoid and its qualities merge with those of collagen and cartilaginous matrix.
It has not seemed practical to divide the osteosarcomas into sclerotic and lytic subtypes, but some special types, such as periosteal, telangiectatic, and low-grade central, have special features that will be elaborated. Although most osteosarcomas are of unknown cause, some sarcomas have Paget disease as a precursor, especially those in older patients. Of the 1,952 osteosarcomas in the Mayo Clinic series, 61 arose in pagetic bone, as did 7 fibrosarcomas, 3 malignant fibrous histiocytomas, 1 giant cell tumor, and 1 malignant lymphoma.
An increasing number of sarcomas occurring after radiation therapy of bone are being recognized. There were 110 postradiation osteosarcomas in this series. Other lesions that arose in irradiated bone were 48 fibrosarcomas, 12 malignant fibrous histiocytomas, 5 chondrosarcomas, 2 angiosarcomas, 1 malignant lymphoma, and 1 Ewing tumor.
Dedifferentiated chondrosarcoma with foci of osteosarcoma are described in Chapter 6. Of the 145 dedifferentiated chondrosarcomas, 80 had a dedifferentiated component that was considered to be an osteosarcoma. Osteosarcoma of the jaw has special features to be described.
A special type of osteosarcoma that grows slowly, metastasizes late (if at all), and is characteristically juxtacortical or parosteal in location is known as parosteal osteosarcoma. It is discussed in Chapter 12.
Extraskeletal osteosarcomas occur in older adults, are almost always high grade, and are associated with a poor prognosis. They are excluded from the series discussed here.
The cause of osteosarcoma is unknown. As indicated above, Paget disease and previous irradiation are known to be associated with a higher incidence of osteosarcoma. It has been suggested that preceding trauma may contribute to the causation of bone tumors. In the Mayo Clinic series, there was only a single well-documented example of previous trauma associated with later development of osteosarcoma. This case involved a 37-year-old man who incurred a bullet wound to the leg 11 years before an osteosarcoma developed at exactly the same site. Whether the trauma or the fragments of lead were related to the later development of osteosarcoma is unknown. Brien and coauthors reported an example of an osteosarcoma arising in the site of previous total hip arthroplasty. This case also suggests that metallic ions may predispose to the development of osteosarcoma.
Evidence has been accumulating that at least some cases of osteosarcoma may be related to a genetic abnormality. It has been well recognized that patients with the hereditary form of bilateral retinoblastoma are at high risk for the development of osteosarcoma. In the Mayo Clinic series, there were only two such patients. Benedict and coauthors found that a suppressor gene (located on chromosome 13) is lost in patients with retinoblastoma and osteosarcoma. Beigel and coauthors studied the karyotypes of several osteosarcomas and found complex abnormalities, but there was a consistent loss of normal chromosome 13 homologue in all cases studied. This, again, suggests a relation with the retinoblastoma gene. In the Mayo Clinic series, multicentric osteosarcoma developed in two brothers with Bloom syndrome. Two siblings with Rothmund-Thomson syndrome had osteosarcoma. One had multicentric osteosarcoma, and the other had a solitary lesion. A third patient probably had the syndrome. One patient had Li-Fraumeni syndrome. Another patient had multiple metachronous osteosarcoma, which she survived, but she died later with bilateral breast carcinoma. This patient’s daughter had a rhabdomyosarcoma of the temporal region, suggesting that this patient may also have a genetic syndrome. One patient with multiple osteosarcomas had preexisting osteopoikilosis. In one patient with osteosarcoma, Ewing sarcoma had been diagnosed 6 years previously at the same site treated only with chemotherapy.
INCIDENCE
The 1,952 osteosarcomas (excluding the parosteal variety) accounted for 27.5% of all malignant tumors and 19.2% of all bone tumors. Osteosarcoma is by far the most common malignant bone tumor (excluding myelomas diagnosed with bone marrow biopsy).
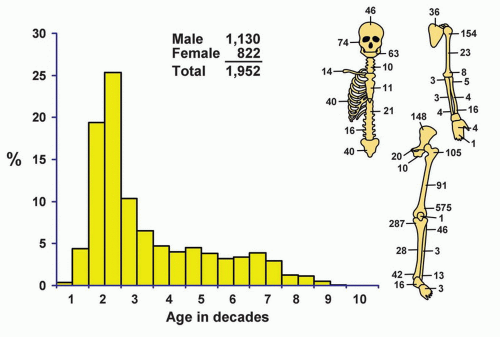 Figure 11.1. Distribution of osteosarcomas according to age and sex of the patient and site of the lesion. |
SEX
Approximately 58% of the patients with osteosarcoma were male. Of the 137 patients with osteosarcoma of the jaws, 55% were male.
AGE
Although a few patients with osteosarcomas are in the first decade of life, the peak incidence is in the second decade (44.77%), and there is a steady, gradual decrease thereafter (Figs. 11.1 & 11.2). Eight patients were younger than 5 years, and the youngest patient was 2 years 11 months old. Six of these eight very young patients were girls. One hundred ninety-two patients were older than 60 years (Fig. 11.3). Of these, 99 were male and 93 were female. Of the 192 older patients, 59 had a preexisting condition: Paget disease (32), previous radiation (24), infarct (1), chronic osteomyelitis (1), and cyst of degenerative joint disease (1). The last condition probably should be considered coincidental. Data from Memorial Sloan-Kettering Cancer Center in New York also pointed to the high likelihood of a secondary osteosarcoma in an older age group.
LOCALIZATION
The metaphyseal part of the long bones is the site of predilection, and almost one-half of the osteosarcomas in the Mayo Clinic series were in the region of the knee.
Of the total number of osteosarcomas, only 24 were distal to the ankle and wrist joints. One patient developed an osteosarcoma in a phalanx of the hand; none had a tumor of the phalanges of the foot. Mirra and coauthors found only one example of an osteosarcoma of the phalanx of a toe in 4,214 cases of conventional osteosarcoma. Sarcomas that did not extend to within 5 cm of an articular surface of a long bone were considered to be in its midportion. Of the 1,430 osteosarcomas involving the long bones, 152 (10.62%) were considered diaphyseal.
Of the total number of osteosarcomas, only 24 were distal to the ankle and wrist joints. One patient developed an osteosarcoma in a phalanx of the hand; none had a tumor of the phalanges of the foot. Mirra and coauthors found only one example of an osteosarcoma of the phalanx of a toe in 4,214 cases of conventional osteosarcoma. Sarcomas that did not extend to within 5 cm of an articular surface of a long bone were considered to be in its midportion. Of the 1,430 osteosarcomas involving the long bones, 152 (10.62%) were considered diaphyseal.
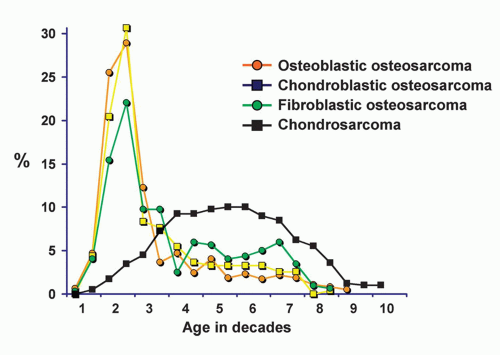 Figure 11.2. Age distribution of patients with chondrosarcoma compared with the distributions in chondroblastic, fibroblastic, and osteoblastic osteosarcoma. Note that all types of osteosarcomas tend to occur in the first two decades of life, whereas chondrosarcomas occur predominantly in adults. |
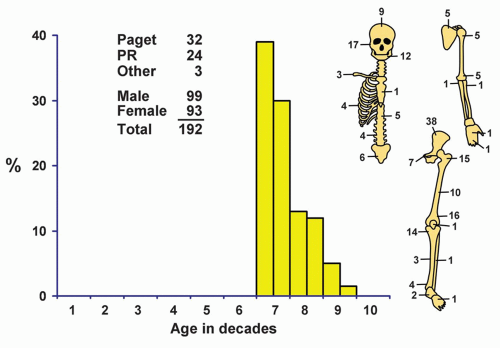 Figure 11.3. Distribution of osteosarcomas in patients 60 and older according to age and sex of the patient and site of the lesion. PR, postradiation. |
Excluding osteosarcoma of the jaws, 77.4% of the tumors arose in the long tubular bones. However, in patients older than 60, only 39% of the tumors arose in long tubular bones. Huvos also noted that in the Memorial Sloan-Kettering Cancer Center group of older patients with osteosarcoma, the axial skeleton was the most common site.
SYMPTOMS
Pain, which initially may be intermittent, and swelling are the cardinal symptoms. Because they are nonspecific, one should not ignore the possible seriousness of these complaints, especially when they occur in children, adolescents, or young adults. It is extremely uncommon for patients with osteosarcoma to be asymptomatic. One patient with osteosarcoma of the femur
presented after a football injury. He had no symptoms before the injury. Pathologic fracture is uncommon. In the Mayo Clinic series, two osteosarcomas of the femur were associated with old infarcts, and two tumors developed in chronic osteomyelitis: one in the femur and one in the tibia. One patient with osteosarcoma had associated myasthenia gravis, and one patient with osteosarcoma of the tibia had radiographic evidence of rickets (oncogenic osteomalacia). Cheng and coauthors also reported on a patient with osteosarcoma associated with oncogenic osteomalacia. The duration of symptoms preceding definitive therapy varies from a few weeks to several months. A history of trouble for more than 1 year is uncommon in patients with conventional osteosarcoma. Swelling or an increase in pain suggests malignant change in Paget disease. Similarly, a flare-up of symptoms in a patient who had irradiation for a benign condition of bone should arouse suspicion. Also, rapid progression of disease is an ominous sign in a patient with known or suspected cartilaginous tumors.
presented after a football injury. He had no symptoms before the injury. Pathologic fracture is uncommon. In the Mayo Clinic series, two osteosarcomas of the femur were associated with old infarcts, and two tumors developed in chronic osteomyelitis: one in the femur and one in the tibia. One patient with osteosarcoma had associated myasthenia gravis, and one patient with osteosarcoma of the tibia had radiographic evidence of rickets (oncogenic osteomalacia). Cheng and coauthors also reported on a patient with osteosarcoma associated with oncogenic osteomalacia. The duration of symptoms preceding definitive therapy varies from a few weeks to several months. A history of trouble for more than 1 year is uncommon in patients with conventional osteosarcoma. Swelling or an increase in pain suggests malignant change in Paget disease. Similarly, a flare-up of symptoms in a patient who had irradiation for a benign condition of bone should arouse suspicion. Also, rapid progression of disease is an ominous sign in a patient with known or suspected cartilaginous tumors.
An increased level of alkaline phosphatase, which occurs in about half the patients, reflects osteoblastic activity.
PHYSICAL FINDINGS
A painful mass in the affected region is usually apparent. If the mass is very large, it may be associated with overlying prominent veins and even edema distal to the lesion. Physical examination is noncontributory in some patients with tumors covered by a thick layer of tissue. Evidence of pathologic fracture is distinctly uncommon.
Some osteosarcomas are familial, and some are associated with generalized skeletal disease, such as osteogenesis imperfecta.
RADIOGRAPHIC FEATURES
The radiographic appearance varies greatly depending on the amount of ossification and calcification in the osteosarcoma. Tumors may be completely lytic or predominantly sclerotic, but they usually have a combination of these features. The destructive process may be limited to the medulla, but it usually involves the cortex as well, and the cortex is nearly always perforated by the growing tumor. Because of a gradual transition from zones of pronounced lysis to zones of uninvolved bone, the borders of the lesion are indistinct. Nonneoplastic bone is deposited, sometimes in layers, when the periosteum is elevated by the perforating tumor (Codman triangle). With continued development of the neoplasm, a large soft-tissue mass is frequently seen contiguous to the bone (Figs. 11.4, 11.5, 11.6, 11.7, 11.8, 11.9, 11.10, 11.11 and 11.12).
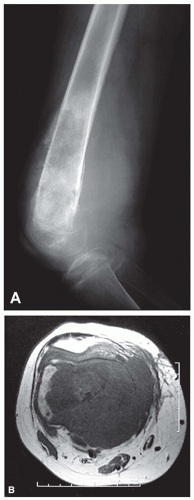 Figure 11.4. Primary high-grade osteosarcoma. Lateral radiograph (A) and axial T1-weighted magnetic resonance image (B) of the distal femur show the classic features of a primary high-grade osteosarcoma with a large mixed lytic and sclerotic destructive lesion involving the distal femoral metadiaphysis, with cortical destruction, extensive malignant periosteal new bone formation, and a large soft-tissue mass. As seen in (B), the popliteal artery and vein are displaced but not encased by the mass. |
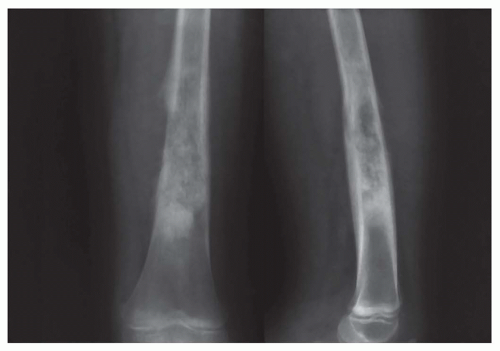 Figure 11.5. Anteroposterior (left) and lateral (right) views of an osteosarcoma involving the shaft of the femur. Approximately 10% of osteosarcomas involve the shaft. The tumor has an aggressive radiographic appearance, with cortical destruction and periosteal reaction with Codman triangle. |
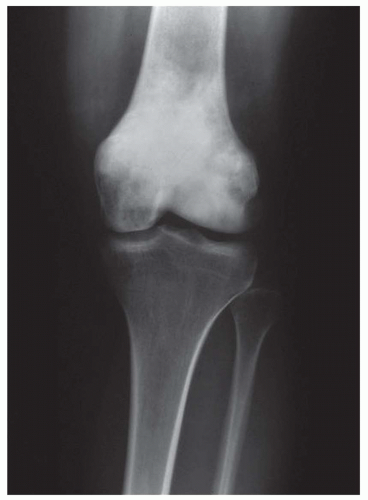 Figure 11.6. Osteosarcoma forming a densely sclerotic mass in the distal femur. |
When the osteosarcoma produces calcifying and ossifying osteoid substance, various degrees of density are seen within the affected portion of the bone. These densities often extend into the contiguous soft tissues. The proliferated bone produced by the neoplastic cells characteristically has a “cloudlike” appearance and ill-defined margins. Usually, the radiographic diagnosis is
easily made when destruction of bone is combined with proliferation of new bone, but definitive therapy should never be initiated without confirmation by biopsy. Some osteosarcomas may be deceptively benign-appearing; some even resemble cysts of bone.
easily made when destruction of bone is combined with proliferation of new bone, but definitive therapy should never be initiated without confirmation by biopsy. Some osteosarcomas may be deceptively benign-appearing; some even resemble cysts of bone.
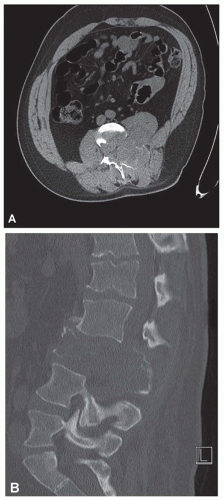 Figure 11.7. Axial (A) and two-dimensional sagittal (B) computed tomographic images of the lumbar spine show a large destructive lytic lesion involving the body and posterior elements of the third lumbar vertebra, with associated cortical destruction and a soft-tissue mass. Although the lesion is predominantly lytic, there is suggestion on the axial images of subtle hazy matrix production within the posterior aspect of the lesion. There is an expansile component to the lesion in the posterior elements. |
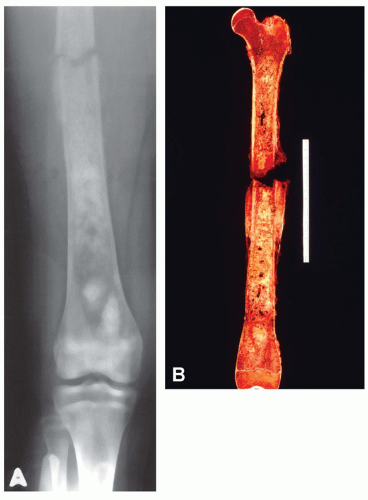 Figure 11.8. Extensive osteosarcoma involving almost the entire femur in a 14-year-old girl. A: The patient was completely asymptomatic until a pathologic fracture developed. B: The gross specimen after disarticulation at the hip shows that the tumor grows from near the greater trochanter almost down to the articular cartilage of the distal femur. |
Osteoid substance, even if present in large amounts as in an osteoblastic osteosarcoma, does not produce radiodensity if it is completely uncalcified. Generally, however, a very sclerotic osteosarcoma is usually, but not necessarily, osteoblastic.
The use of plain radiographs is the most effective way of localizing and suggesting a diagnosis of osteosarcoma. A radioactive isotope bone scan may be helpful in demonstrating multicentric osteosarcoma. Modern imaging techniques, such as computed tomography and magnetic resonance imaging, are routinely used for a preoperative staging study in patients with osteosarcoma. With the widespread use of limb-sparing surgery, accurate pretreatment staging of these tumors has become even more important. McLeod and Berquist emphasized that although the use of plain radiographs is the standard means for diagnosing osteosarcoma, computed tomograms and magnetic resonance images are far superior in delineating the extent of the disease. Computed tomograms may be superior in axial locations, but magnetic resonance images are superior in the extremities. Both T1- and T2-weighted sequences are necessary to accurately delineate the intramedullary and extraosseous extent of osteosarcomas on magnetic resonance imaging sequences. Normal marrow is “bright” on T1-weighted images and “dark” on T2-weighted images. In contrast, osteosarcomas are “dark” on T1-weighted images and “bright” on T2-weighted images. These contrasting signals help to accurately delineate the extent of the tumor. These images also help in determining if the neurovascular bundle is involved. Redmond and coauthors and Gillespy and coauthors also emphasized the superiority of magnetic resonance imaging studies for preoperative staging of osteosarcoma.
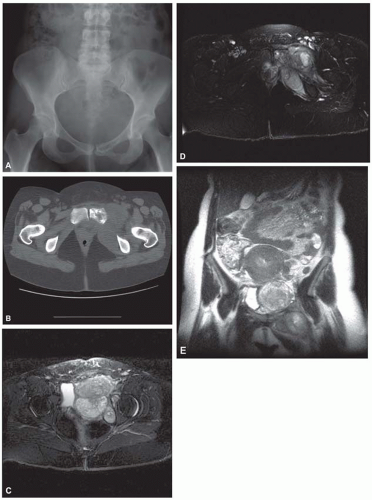 Figure 11.9. Osteosarcoma. Anteroposterior radiograph (A) and axial computed tomogram (B) of the pelvis in a 33-year-old woman show a mixed lytic and sclerotic destructive lesion involving the left pubic bone with a large associated unmineralized soft-tissue mass. Axial (C and D) and coronal (E) T2-weighted magnetic resonance images show the anatomical extent of the soft-tissue mass, which has both an intrapelvic and extrapelvic component. The soft-tissue mass fills the left hemipelvis, where it causes significant mass effect on the bladder and extends along the pelvic sidewall through the obturator foramen to the adductor region of the proximal thigh. |
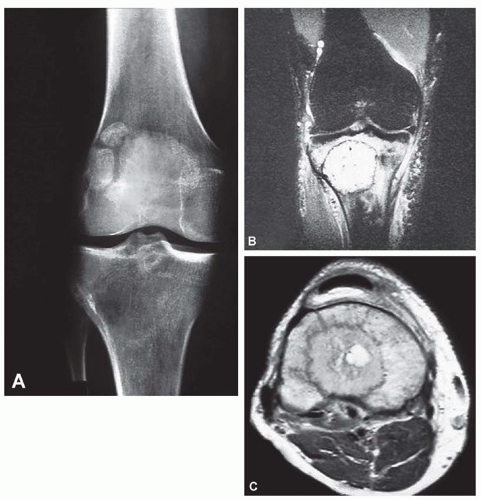 Figure 11.10. Osteoblastic grade 4 osteosarcoma involving the proximal tibia. A: Anteroposterior radiograph shows a tumor with indeterminate findings. B and C: Magnetic resonance imaging is helpful in demonstrating more clearly the aggressive features that suggest malignancy. |
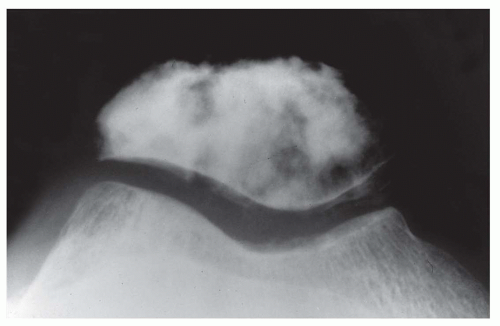 Figure 11.11. Osteosarcoma forming a sclerotic mass in the patella, a very uncommon location for osteosarcoma. |
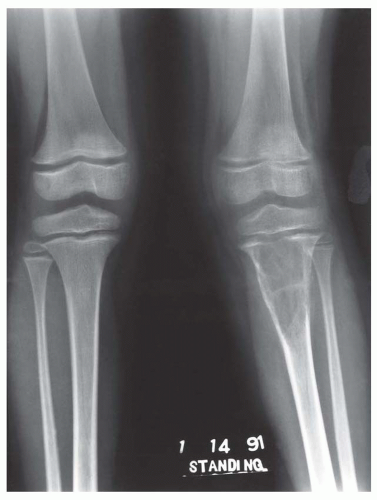 Figure 11.12. Osteosarcoma involving the proximal tibia in a young girl. The lesion was associated with hyperphosphatemic osteomalacia, as manifested by the widened epiphyseal plates. This appearance changed after the patient was treated with chemotherapy. |
Pulmonary metastasis is sometimes found when the patient first seeks medical advice. Computed tomography aids in demonstrating pulmonary metastasis. In a small percentage of patients, computed tomograms reveal pulmonary metastasis when plain radiographs are negative, and they may show more disease than is obvious on radiographs.
GROSS PATHOLOGIC FEATURES
By the time a patient receives definitive therapy, the osteosarcoma has generally breached the cortex. The extraosseous mass may even completely encircle the bone. The periosteum presents a barrier that often becomes greatly distended before it is perforated. Similarly, the epiphyseal plate acts as a relative barrier to the growth of osteosarcoma. Cortical destruction that is slight to complete is found at the site of perforation (Figs. 11.13 & 11.14).
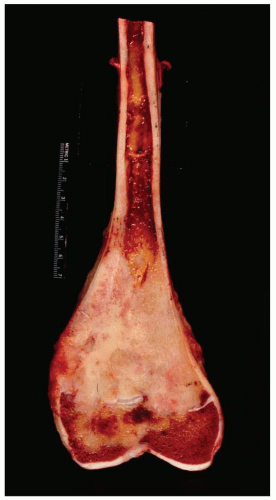 Figure 11.13. Typical gross appearance of osteosarcoma involving the distal femoral metaphysis. The tumor has destroyed the epiphyseal plate and extends almost down to articular cartilage. There is also a soft-tissue mass. |
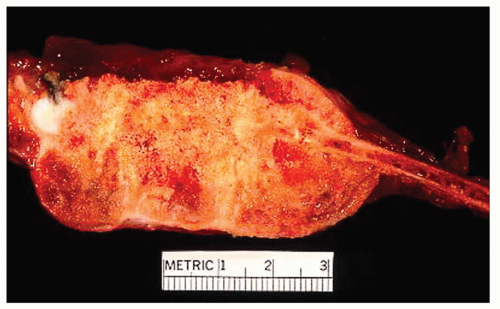 Figure 11.14. Osteosarcoma arising from the second rib in a 21-year-old woman, who complained of chest pain for 3 years. The tumor extends beyond the bone to create a large soft-tissue mass. |
Some of the tumors spread in the marrow cavity for unexpectedly great distances, occasionally beyond the area visible on the radiograph, and this spread must be considered during therapy. In nearly all instances, the extent of marrow involvement is readily apparent grossly when the bone is sawed longitudinally, and most tumors do not spread in the marrow beyond their gross extraosseous limits. Skip areas of medullary involvement are extremely rare, although Enneking and Kagan stressed their importance. With modern imaging techniques, the clinician is unlikely to miss a rare skip metastatic lesion.
Nearly all osteosarcomas have such a prominent central component that a central origin is logically assumed. In the Mayo Clinic series, none of the osteosarcomas seemed to arise within the cortex. Rarely, however, highly malignant tumors were located mainly outside the bone and involved only the outer portion of the cortex, findings suggestive of a periosteal origin.
As indicated by the radiographic findings, osteosarcomas vary from extremely soft, fleshy masses through a firm, fibrous tumor with foci of irregular ossification and various amounts of chondroid material to a densely sclerotic type (Figs. 11.15, 11.16 and 11.17). Sclerosis, when present, is invariably most pronounced in the central regions. Nearly all osteosarcomas, however sclerotic,
have soft peripheral zones that can be sectioned without preliminary decalcification. Most osteosarcomas have a soft tissue component by the time a diagnosis is made, so that the surgeon need not breach the cortex to get tissue from within the medullary cavity for diagnostic purposes. Areas of necrosis, cyst formation, telangiectasis, and hemorrhage are most likely to occur in the soft tumors.
have soft peripheral zones that can be sectioned without preliminary decalcification. Most osteosarcomas have a soft tissue component by the time a diagnosis is made, so that the surgeon need not breach the cortex to get tissue from within the medullary cavity for diagnostic purposes. Areas of necrosis, cyst formation, telangiectasis, and hemorrhage are most likely to occur in the soft tumors.
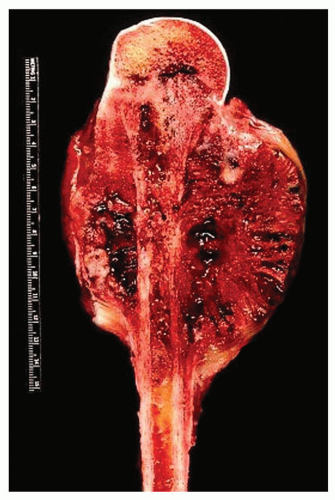 Figure 11.15. Fibroblastic grade 4 osteosarcoma involving the proximal humerus in a 13-year-old boy. The tumor forms a massive, partially hemorrhagic, and necrotic soft-tissue mass. Treatment included amputation without adjuvant therapy. The osteosarcoma was cured; however, 17 years later an oligodendroglioma developed. The patient died 7 years later of recurrent glioma. |
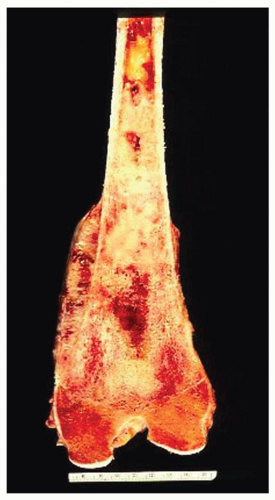 Figure 11.16. No gross features suggest that this osteosarcoma has a cartilaginous component. However, the microscopic features were those of a chondroblastic grade 3 osteosarcoma. |
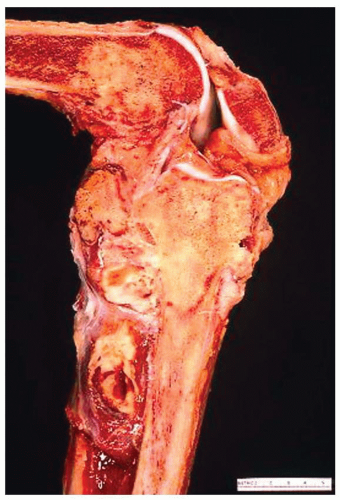 Figure 11.17. Amputation specimen after chemotherapy for an extensive osteosarcoma involving the proximal tibia. Tumor involves the distal femur and the proximal tibia, and there are extensive soft-tissue masses. All the tumor was necrotic. |
Metastasis is predominantly hematogenous, with the production of pulmonary deposits. Metastasis to other bones may be early and widespread, suggesting a multifocal origin of the sarcoma, or delayed and localized, suggesting that a new tumor has developed. In 42 patients in the Mayo Clinic series, more than one bone was involved with osteosarcoma. Twenty-six of the 42 patients had metachronous osteosarcoma. The interval between the two sarcomas ranged from 9 months to 14 years. The interval was less than 1 year in 5 patients, between 1 and 2 years in 7, between 2 and 5 years in 9, between 5 and 10 years in 3, and more than 10 years in 2. One of the patients had Paget disease, and one other patient probably had Rothmund-Thomson syndrome but the evidence was not conclusive. Five patients were cured of their tumor; one of these patients died of bilateral breast carcinoma 15 years later. This patient’s daughter developed embryonal rhabdomyosarcoma of the temporal region.
Sixteen patients had synchronous osteosarcoma, four of whom had multiple skeletal sites of involvement. Five of the 16 patients had preexisting conditions: Rothmund-Thomson syndrome (1), Bloom syndrome (1), Li-Fraumeni syndrome (1), Paget disease (1), and osteopoikilosis (1). Only one patient survived the sarcomas, but this patient developed another osteosarcoma at 10 years and died of that tumor.
Most patients with osteosarcoma receive preoperative chemotherapy. The material sent to the laboratory usually is a resection specimen rather than an amputation specimen. However, the handling of the specimen is essentially the same. At Mayo Clinic, the surgeon usually sends a specimen of the marrow from the resection margin so it can be checked on frozen section. The gross specimen then should be stripped of all extraneous soft tissue, so that only the affected bone and tumor remain. Raymond and Ayala described the techniques for the gross examination of postchemotherapy specimens. They attempt to cut the specimen where preoperative angiograms suggest the most viable tumor will be. How many sections of the specimen should be taken is a question that is still not settled. At Mayo Clinic, we make one longitudinal section through the middle of the specimen. Using a band saw, we then obtain a thin slice of the entire specimen. This specimen is put in a plastic bag and “photographed” with a xerographic copier. The copy serves as a template for mapping of the specimen. The entire slab is decalcified, a process requiring several days because of cortical bone. After decalcification is complete, the entire specimen is cut into blocks and labeled according to the xerographic figure of the gross specimen.
HISTOPATHOLOGIC FEATURES
The histopathologic features of osteosarcoma vary greatly, as mentioned above. Lichtenstein tersely stated the essential criteria as “(1) the presence of a frankly sarcomatous stroma and (2) the direct formation of tumor osteoid and bone by this malignant connective tissue.”
Although osteosarcomas can be divided rather conveniently into osteoblastic, chondroblastic, and fibroblastic groups, depending on the dominant histologic pattern, it is necessary to be arbitrary in some cases. Occasionally, a highly anaplastic tumor contains no osteoid but is otherwise so similar to an osteoid-producing tumor in histologic appearance that it logically must be classified as an osteoblastic sarcoma. Some such tumors have features that qualify them to be malignant fibrous histiocytomas. However, some of these tumors show production of matrix in foci or occasionally in metastatic tumors. This problem has been highlighted by Balance and coauthors, who use the term osteogenic sarcoma, malignant fibrous histiocytoma subtype. Other tumors that seem to be nearly pure fibrosarcomas contain foci of homogeneous, afibrillar, eosinophilic material that resembles hyalinized collagen. When such foci cannot be differentiated with certainty from osteoid tissue, the tumor containing these foci is best classified as fibroblastic osteosarcoma. An occasional fibroblastic tumor with only questionable osteoid production produces very sclerotic metastases. The usual member of this group, however, contains obvious osteoid material (Figs. 11.18, 11.19, 11.20, 11.21, 11.22, 11.23, 11.24, 11.25, 11.26, 11.27, 11.28, 11.29, 11.30, 11.31, 11.32, 11.33 and 11.34).
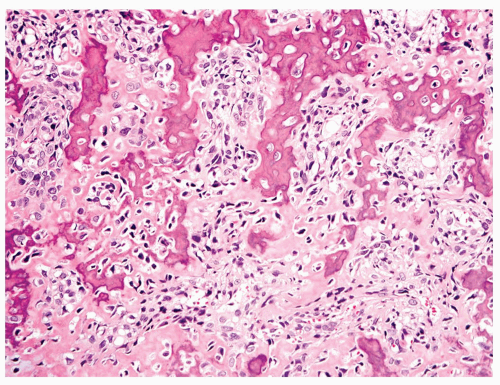 Figure 11.18. Osteoblastic grade 4 osteosarcoma. The tumor contains abundant osteoid intimately associated with anaplastic tumor cells. |
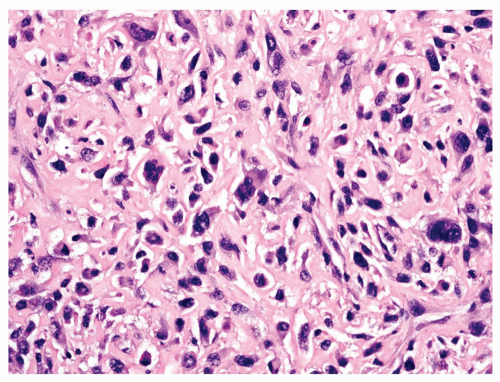 Figure 11.19. Osteosarcoma with highly atypical spindle cells. Matrix is present between the tumor cells. |
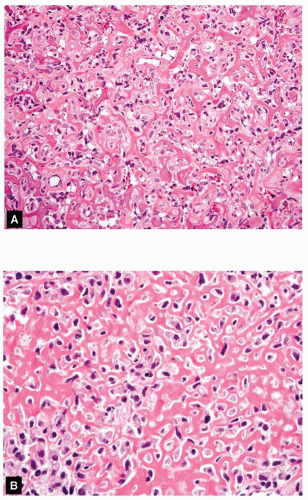 Figure 11.20. Low- (A) and high- (B) power views of an osteoblastic grade 4 osteosarcoma show a lacelike pattern of osteoid production. |
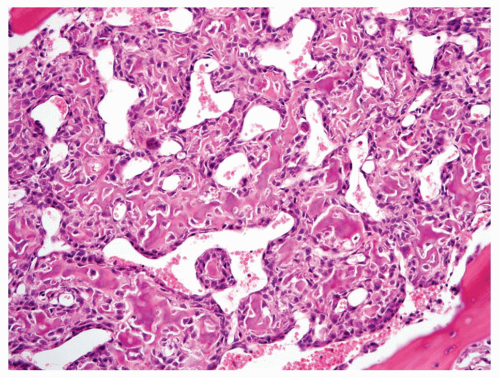 Figure 11.21. An example of osteoblastic osteosarcoma with a hemangiopericytomatous vascular pattern. |
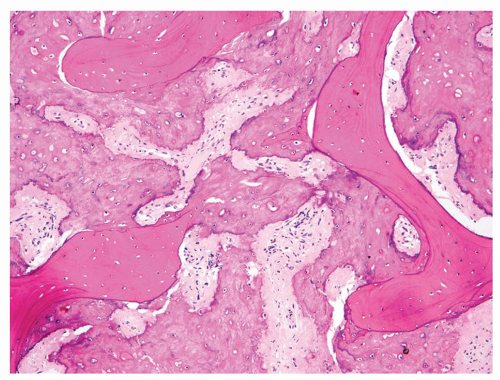 Figure 11.22. Sclerotic osteosarcoma. The tumor has produced so much matrix that the tumor cells have been markedly compressed. A diagnostic feature of malignancy is that the tumor permeates between preexisting trabeculae of medullary bone. |
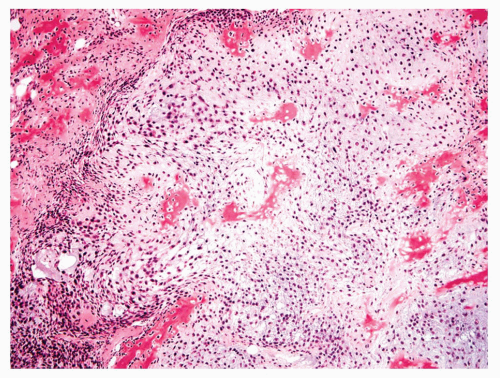 Figure 11.23. Typical chondroblastic osteosarcoma. The cartilage has malignant-appearing cells in lacunae, and there is crowding at the periphery of the lobule where sheets of spindle cells are formed. Islands of osteoid are seen within the cartilage. |
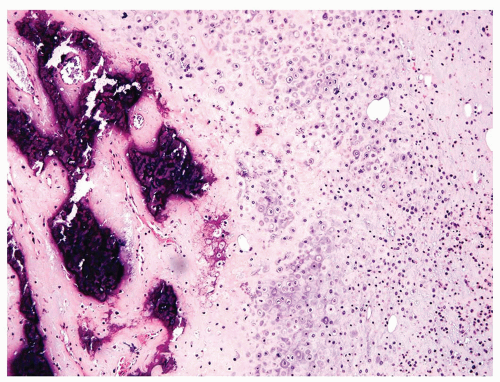 Figure 11.24. This chondroblastic osteosarcoma contains sheets of grade 3 cartilage that merge into large areas of mineralized osteoid. |
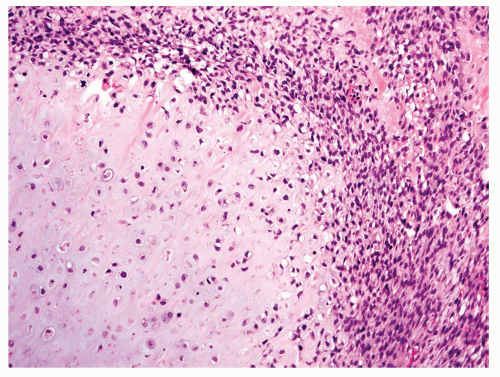 Figure 11.25. An example of chondroblastic grade 4 osteosarcoma containing malignant cartilage, bone, and high-grade spindle cells. |
Approximately 56% of osteosarcomas in the Mayo Clinic series can be classified as osteoblastic. Most commonly in this group, osteoid is present as a fine lacelike network between individual tumor cells. The tumor cells have obvious features of malignancy, such as nuclear hyperchromasia and abundant mitotic activity, including atypical mitotic figures. The matrix may undergo calcification focally (Figs. 11.18, 11.19, 11.20 and 11.21). Occasionally, the matrix is produced in the form of bony trabeculae rather than osteoid. The trabeculae are usually thin and anastomosing. Rarely, thick, well-formed bony trabeculae are seen in an otherwise high-grade osteosarcoma. Some osteoblastic osteosarcomas are extremely sclerotic. The sclerosis may be so prominent that the tumor cells are not visible. In this instance, the matrix completely fills up the marrow cavity and entraps preexisting bony trabeculae. The diagnosis of these extremely sclerotic osteosarcomas may have to be made only on the basis of permeation and without identifying malignant cells (Fig. 11.22).
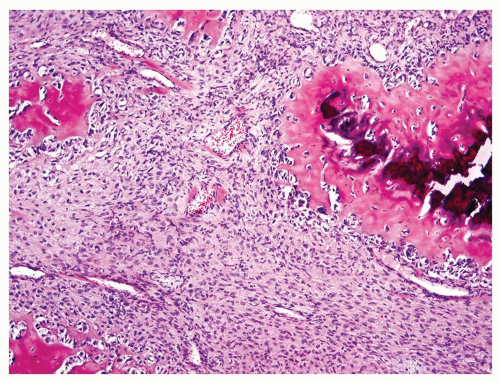 Figure 11.26. Fibroblastic grade 3 osteosarcoma. Irregularly shaped nodules of osteoid are surrounded by malignant spindle cells. |
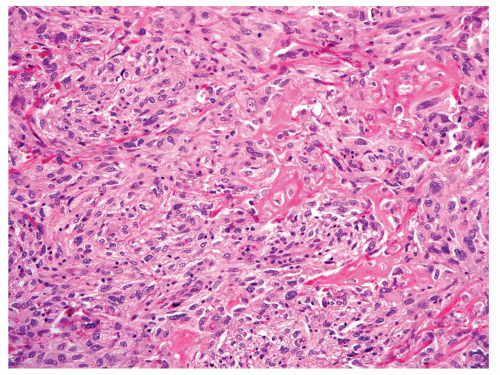 Figure 11.27. Fibroblastic grade 4 osteosarcoma. Malignant osteoid production is present within fascicles of atypical spindle cells. |
Approximately 20% of the osteosarcomas in this series were classified as chondroblastic osteosarcoma. The cells lie in lacunae and form lobules. The cytologic features of the cells in lacunae are very similar to the cytologic features of spindling tumors seen elsewhere in the neoplasm. The center of the chondroid lobule frequently has bony trabeculae that produce a feathery appearance. Toward the periphery of the lobule, the tumor becomes hypercellular and sheets of spindle cells are seen. Osteoid matrix is usually present between the tumor cells in the spindling areas (Figs. 11.23, 11.24 and 11.25). Occasionally, a chondroblastic tumor has extensive spindling without clear-cut osteoid production. These tumors are logically classified as chondroblastic osteosarcoma. The lack of osteoid is assumed to be a function of sampling.
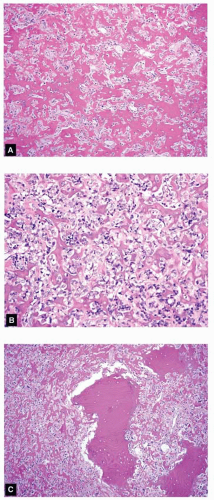 Figure 11.28. A: Areas of this osteoblastic osteosarcoma involving the transverse process of the first thoracic vertebra simulate the appearance of osteoblastoma. The radiographic images showed the features typical of osteoblastoma. B: However, obvious cytologic atypia was evident in other microscopic fields. C: Destructive permeation of preexisting trabeculae of bone was also present. |
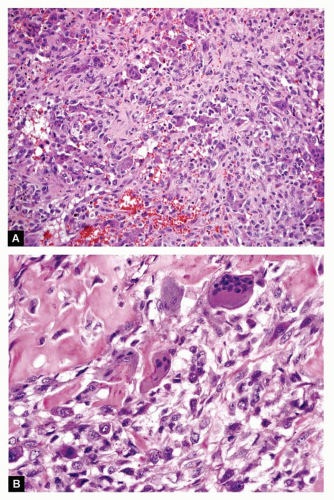 Figure 11.29.
Get Clinical Tree app for offline access
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree


|





