10 ORTHOPEDICS AND MUSCULOSKELETAL CONDITIONS Kevin P. Murphy, Colleen A. Wunderlich, Elaine L. Pico, Sherilyn Whateley Driscoll, Elizabeth Moberg Wolff, Melanie Rak, and Maureen R. Nelson GROWTH AND DEVELOPMENT OF THE BONY SKELETON The skeletal system develops from mesoderm and neural crest cells (1). Somites form from paraxial mesoderm and differentiate into sclerotomes, dermatomes, and myotomes. Sclerotome cells migrate from the somite and ultimately become chondrocytes. The remaining dermatome cells form the dermis. Myotome cells give rise to striated muscles of the backs of limbs (Figure 10.1). Limbs and respective girdles, the appendicular skeleton, are derived from cells of the lateral plate mesoderm. Limb buds appear in utero approximately on day 26 for the upper extremities and day 28 for the lower extremities (2). The hand plate forms in the fifth week, with digitization of rays in the sixth week. Notches appear between the rays in the seventh week, the failure of which results in syndactylism. During the seventh week, the limbs also rotate laterally in the upper extremities and medially in the lower extremities. This brings the thumb to the more lateral position in the upper extremity and the great toe to the more medial position in the lower extremity. Chondrification begins in the sixth week, followed by early ossification in the seventh week and subsequent joint cavity formation in the sixteenth week. By the eighth week, definite muscle formation is noted, as the embryo assumes a human appearance and basic organ systems are completed. The fetal period begins at 9 weeks with rapid growth and changes in body proportion (3). Knowledge of the normal proportions and growth and development of the musculoskeletal system allows a firm foundation for the understanding of both congenital and acquired conditions requiring care in the developmental years. Figure 10.2 displays the growth rates for boys and girls by age. About half of the individual’s height is reached by age 2 and three-fourths by age 9. Prediction of adult height can be obtained by plotting bone age against current height to determine percentile value (Figure 10.3). Following the percentile to skeletal maturation is a method for estimating final adult height. Paley height multipliers offer an even simpler way of estimating adult height at any child age (4). Predictions are less accurate for the younger child (Figure 10.4). The reader is referred to more detailed references for tables displaying differences over time and growth and rates for standing, sitting, and subischial lengths in boys and girls (5). The measurement of arm span provides an indirect control parameter for the measurement of standing height, particularly useful in those who are nonambulatory. To measure arm span, the patient simply raises the arms to a horizontal position, and the distance between the tips of the middle fingers is measured with a tape measure (6,7). The standing height is about 97% of arm span. In children with spinal deformity, arm span is a good estimate of what standing height would be if there were no abnormal curvatures. It is well known that different proportions of the body grow and change at different percentages over the developmental years (Figure 10.5). CONGENITAL CONDITIONS Minor limb deficiencies are relatively common in the upper and lower extremities. Syndactyly occurs in approximately 1 in 2,200 births, either as cutaneous with simple webbing of the fingers or osseous with fusion of the bones when the digital rays fail to separate between the fifth to eighth weeks of gestation (8). It is most frequent between the third and fourth fingers and between the second and third toes, and is inherited as a simple dominant or simple recessive trait. It can occur in isolation or as part of a syndromic condition. Surgical separation of the digits is more common with complete syndactyly for functional and cosmetic reasons. Polydactyly has an incidence of approximately 1 to 1.5 per 1,000 live births and is the most common congenital toe deformity (5). Eighty percent of polydactyly in the foot occurs with the fifth toe. Most often an isolated trait, an autosomal-dominant inheritance pattern has been identified with variable expressivity. Radiographic evaluation is necessary to define duplicated structures. Deferring radiography until after 6 months of age allows phalanges to ossify. Surgery around the age of 1 not only improves cosmesis, but also is helpful in facilitating shoe fitting. FIGURE 10.1 Trilaminar disk. Neural tube closure. Mesoderm differentiates into dermatome, myotome, and sclerotome. Migrating sclerotome cells become chondrocytes. Chondrocytes ultimately form vertebral bodies and arches. Camptodactyly, translated from Greek, means “bent finger.” The proximal interphalangeal (PIP) joint is flexed, most commonly digit 5. Incidence is felt to be less than 1% of the general population with equal gender distribution (8,9). Appearance in adolescence, often girls, is less common. Surgical reconstructions are for functional and cosmetic reasons. Malformations of the radius are more common than those of the ulna and are associated with numerous syndromes (5,10,11,12). In children with limb anomalies, a multisystemic review is generally indicated because abnormalities in other systems are often present. Simple and multifactorial inheritance may all be causative in addition to teratogenic effects, such as maternal exposure to viral infections and chemical dependency such as alcohol (10). A failure of the scapular to descend from its cervical region overlying the first through fifth ribs results in Sprengel’s deformity (6,13–16). Children often present with a shortened neckline (Figure 10.6). Lack of normal scapulothoracic motion and malpositioning of the glenoid causes limited forward flexion and abduction of the shoulder. An omovertebral bar is present in up to 50% of cases (17). The bar connects between the superior medial angle of the scapula and the cervical spine, and consists of fibrous cartilaginous tissue or bone. It is not uncommon to see other abnormal regional anatomy and syndromes that need to be screened for carefully, including scoliosis, spina bifida, rib anomalies, and Klippel–Feil syndrome (18,19). Renal and pulmonary disorders can also be present, and a renal ultrasound, if not already completed, is indicated. The condition can be bilateral in up to 30% of cases. FIGURE 10.2 Greene and Anderson growth curve. Source: Greene W, Anderson M. Skeletal age and the control of bone growth. Instr Lect Am Acad Orthop Surg. 1960;17: 199–217. FIGURE 10.3 Prediction of adult height. Adult height predicted by plotting child’s bone age versus current height to determine percentile value. Follow the percentile to skeletal maturation for estimate of final adult height. FIGURE 10.4 Paley height multipliers. Charts provide a simple method of predicting adult height for boys and girls. Congenital dislocation of the radial head unaccompanied by other congenital abnormalities of the elbow or forearm is rare (5). Congenital radioulnar synostosis is also a rare condition caused by failure of the radius and ulna to separate, usually proximal (Figure 10.7). The forearm is usually left in significant pronation with the condition bilateral 80% of the time (20). This condition is also associated with multiple other syndromes, which need to be carefully screened for (10). Children present for evaluation depending upon the degree of functional deficit. Radiographs can be helpful when ossification is present. MRI scans of the proximal radius and ulna can reveal more of a cartilaginous synostosis or a fibrous tether that has not ossified. Children with radioulnar synostosis without functional limitation should be observed. Success with surgery to resect the synostosis is often limited with minimal functional gain (5). Rotational osteotomies for pronation deformities greater than 45 degrees can be helpful. Postoperative compartment syndrome of the forearm needs to be watched for (21,22). FIGURE 10.5 Proportions of the body as they change during growth. (Source: Lowrey GH. Growth and Development of Children. 6th ed. Chicago, IL: MYB; 1973.) In about 5% of humans, there are minor variations in the number or proportions of vertebra (23). Osseous anomalies are felt to account for up to 6% of children who present with signs of torticollis. Individuals with cervical fusion are generally apparent on plain cervical radiographs, including flexion and extension views. The Klippel–Feil syndrome, sometimes called brevicollis, is characterized by short neck, low hairline, and restricted neck movement. Consisting of congenital fusions of the cervical vertebra, its incidence is approximately 0.7% (5,11). Failure of segmentation in the cervical spine most often characterizes the Klippel–Feil syndrome. Patients with Klippel–Feil syndrome or related conditions should have a renal ultrasound and cardiac evaluation (echocardiogram). Contact sports are contraindicated, as are similar, more aggressive activities. FIGURE 10.6 Sprengel’s deformity. FIGURE 10.7 Radial ulnar synostosis. Clubfoot, talipes equinovarus, is a common term used to describe several kinds of ankle or foot deformities present at birth. The foot is generally in equinus, with forefoot and hindfoot varus and severe adduction (Figure 10.8). As the most common birth defect, it carries an incidence ranging from 1:250 to 1:1,000 live births, depending on the population (8). The condition is one of the most treatable of birth defects, often leading to normal or near-normal athletic activities later in life (5). Multifactorial genetic inheritance, along with poorly understood environmental factors, may explain the bulk of etiology. Some clubfoot disorders are transient or apparent in nature and result simply from intrauterine crowding. Other conditions may occur in association with myelodysplasia, arthrogryposis, and particularly hip dislocation. Prenatal ultrasound can be effective in diagnosing intrauterine clubfoot, with no false-negative prediction and a true-positive predictor rate of 83% (8). Recent treatment has focused primarily on the Ponseti technique (25,26,27,28). The range of motion (ROM) should be maintained by passive exercise and therapeutic play, particularly into dorsiflexion and eversion. Persistent deformity into adulthood can result in unstable ankles, lateral sprains, and difficulty with weight-bearing and other gross mobility tasks. Metatarsus adductus can be seen in up to 12% of full-term births (8). Intrauterine crowding or positioning may be causative. Flexibility can be determined by fixing the hindfoot in a neutral position and gently manipulating the mid-foot and forefoot to a more lateral position. Internal tibial torsion may be associated, making the thigh-foot angle worse. Serial casting may be helpful in children under 1 year of age. Careful attention should be given not to place the hindfoot in valgus or create a skew foot deformity. Surgery is rarely indicated, but can be done in the more rigid persistent deformities after the age of 5. Various forms of posterior medial release are available (5). Flat feet or pes planus may be flexible or rigid (5). Flexible pes planus is usually asymptomatic, at least in the early years, and is the most common type found in children. Inexpensive scaphoid pads or medial inserts may help to create more plantigrade weight-bearing in the child, but they do not correct the deformity. Extreme cases, such as in children with hypotonia, may require surgery after the age of 5 years in the form of a calcaneal lengthening once the bony cortices are more solid. Untreated progression may occur with compensatory hallux valgus, planovalgus, and secondary bunion and toe deformities. Pes planovalgus is associated with more active or shortened peroneal musculature, progressing over time, with the development of pain, particularly in later years. Rigid pes planus is a congenital deformity associated with other anomalies in 50% of cases (29). It is caused by failure of the tarsal bones to separate leaving a bony cartilaginous or fibrous bridge or coalition between two or more tarsal bones (30,31). Talocalcaneal coalitions tend to become symptomatic earlier, between 8 and 12 years, whereas calcaneonavicular coalitions are more likely to be symptomatic between 12 and 16 years. Symptoms are insidious with occasional acute arch, ankle, and mid-foot pain. The hindfoot often does not align in its normal varus position on tiptoe maneuvers (5). Patients are predisposed to ankle sprain secondary to the limited subtalar motion, and stress to the subtalar and transverse tarsal joints frequently causes pain. Computed tomography (CT) scans are diagnostic, and initial treatment is conservative with short-leg casting or molded orthosis and rest. If conservative care fails, surgical intervention is usually necessary. With all symptomatic pes planus, accessory navicular bones need to be considered (8). Rigid cavus feet may be associated with metatarsalgia, clawing, and intrinsic muscle atrophy. With a cavus foot, stresses are increased across the joints, along with pressures on bony prominences, muscle strength being required to maintain posture. The result is pain, fatigue, and instability. The cavus foot may be caused by an underlying neurologic condition such as Charcot-Marie-Tooth disease, spinal dysraphism, Friedreich’s ataxia, or spinal tumor. Custom-molded inserts or orthosis may be helpful in providing arch support and decreased pain by relieving pressure off bony prominences and providing a shock-absorber effect. Cavus feet can often run in families, making family history critical. Clinical exam for flexibility with localization of the deformity to the forefoot or hindfoot should be completed. The Coleman block test for determination of hindfoot flexibility can be critical, particularly for any surgical repair in the more rigid and symptomatic deformity (32). Plantar fascia release is standard for all cavus foot procedures (33,34). FIGURE 10.8 Clubfoot deformity. Associated forefoot supination, deep medial crease, and equinovarus of the hindfoot. Congenital vertical talus is exceedingly rare (5). The navicular bone is dislocated dorsolaterally on the head of the talus (Figure 10.9). It is commonly associated with neuromuscular and genetic disorders, including trisomy 13, 14, 15, and 18 (35). Clinical features include a rigid convex plantar surface (rocker bottom) with hindfoot equinus and hypoplastic laterally deviated forefoot. Casting can initially have some benefit for contracted dorsolateral soft tissues, but only as a prelude to surgical intervention. A single-stage procedure is generally the consensus (5) and can involve talectomy, naviculectomy, subtalar arthrodesis, and triple arthrodesis. FIGURE 10.9 Vertical talus. Arthrogryposis multiplex congenita refers to a symptom complex characterized by multiple joint contractures that are present at birth. The clinical literature has delineated as many as 150 entities under this term (8,36). The incidence of arthrogryposis as a whole is approximately 1 per 3,000 live births. Amyoplasia (which literally means no muscle growth) affecting all four limbs is less common, at approximately 1 in 10,000 live births (8). There are many different ways to classify the arthrogrypotic conditions (5,37). A simple way is to divide the contracture syndromes into three different groups (5). Group number 1 involves arthrogryposis multiplex congenita, Larsen syndrome, and more or less total body involvement. Larsen syndrome is a rare condition involving multiple congenital dislocations of large joints, a flat facies, and significant ligamentous laxity (38,39). Patients commonly have abnormal cervical spine segmentation with instability and can be associated with myelopathy. Group number 2 would include the distal arthrogryposis predominantly involving hands and feet. Distal arthrogryposis type II involves the presence of facial findings, whereas type I does not. Freeman–Sheldon syndrome is an example of distal arthrogryposis type II, with a characteristic “whistling face” appearance (40,41). Group number 3 involves the pterygium syndromes. Pterygium comes from the Greek word meaning “little wing.” Pterygiums can be isolated or multiple. Multiple pterygium syndrome is characterized by webbing across every flexion crease in the extremities, most prominently across the popliteal space, elbow, and axilla (42). Popliteal pterygium syndrome has features involving the face, genitals, and knees (43). A popliteal web is usually present bilaterally running from the ischium to calcaneus, resulting in severe knee flexion deformities (Figure 10.10). The diagnosis of arthrogryposis can be suspected with prenatal ultrasound. Absence of fetal movements of distal or proximal joints in combination with polyhydramnios is suggestive (44). The birthing process can be complicated by joint contractures, with neonatal fractures resulting. Perinatal fractures are common and believed to be secondary to hypotonia and rigid joints (45). Therapy should not be initiated in a newborn until such fractures are ruled out (46). Children who survive infantile arthrogryposis often have upper and lower extremity involvement in typical patterns. Common deformities of the upper extremities include adduction; internal rotation contractures of the shoulders; fixed flexion or extension contractures of the elbows, either wrist flexion and ulnar deviation or extension and radial deviation; and thumb-in-palm deformities. In the lower extremities, flexion, abduction, and external hip rotation contractures with unilateral or bilateral dislocations are noted. Bilateral dislocations of the hip are more often left alone, whereas unilateral dislocations, because of scoliosis risk, are more often surgically treated (5). Fixed extension or flexion contractures of the knees are also seen along with severe rigid bilateral clubfeet. In the most severe rigid clubfeet, not correctable with casting and conservative care, a talectomy may be necessary or talar enucleation in association with the posterior medial releases. Extension wedge osteotomies of the distal femur may be necessary to correct flexion contractures of the knee. There is always a well-recognized risk of neurovascular damage, with operative correction of knee flexion contractures needing careful consideration to avoid overstretching of the neurovascular bundle. Shortening osteotomies completed at the same time as the extension wedge osteotomy may minimize these risks. In the absence of degenerative neurologic conditions, individuals with arthrogryposis maintain their strength and ROM over time (5). Surgical and rehabilitation goals are generally centered on self-help skills, such as feeding, toileting, and mobility skills such as standing, walking, and transfers using assistive devices as needed. Surgical procedures of the upper extremity are usually delayed until the child is old enough for a more definitive functional assessment to be completed. If both elbows are involved with extension, surgery to increase flexion may be best done on only one side. FIGURE 10.10 Isolated popliteal pterygium. Outcomes appear better if joint surgery is completed prior to the age of 6 to avoid adaptive intra-articular changes (8). Osteotomies for realignment are usually performed closer to skeletal maturity. Early mobility and avoidance of prolonged casting may result in improved ROM and function postsurgery. Most individuals do not have intellectual impairment or sensory deficits. The children often have a keen natural ability to learn substitution techniques. A strong association between initial feeding difficulties and subsequent language development is known, which should not be misidentified as intellectual deficiency (47). BRACHIAL PLEXUS PALSY Birth brachial plexus injury occurs in between 1 and 2 per 1,000 live births in the United States. Babies with increased birth weights, multiparous mothers, and shoulder dystocia are at the highest risk for brachial plexus palsy (48,49). The most widely described mechanism of action for this is lateral stretch, which is logical secondary to the location of the brachial plexus, the high correlation with shoulder dystocia, and the positioning of the mother and infant (49). It has been described that between 50% and 95% of these infants will recover spontaneously. The overarching goal of treatment of brachial plexus injuries is maximizing arm and hand function. Goals are normalization of limb function, with optimization of nerve regeneration and mechanical increase of elbow flexion and shoulder stabilization. This can be achieved through aggressive rehabilitation and surgical intervention (48). For any nerve that is injured, classification makes evaluation and comparison clearer. The Seddon Classification of Nerve Injury is commonly used. Neurapraxia occurs with no lasting anatomic changes, with fibers preserved. This is exemplified by a football “stinger” injury. Complete resolution is expected. In axonotmesis, there is an interruption of neural continuity to some degree. There is an extremely variable level of deficit that is difficult to evaluate and predict the degree of recovery. Neurotmesis is the most severe injury, with total disruption of the elements of the nerve, and this will not independently recover. If it is preganglionic, or proximal to the dorsal root ganglion, it is called an avulsion. If it is postganglionic, or distal to the dorsal root ganglion, it is called a rupture (50). Both of these require surgical intervention for recovery. There are also descriptors for the levels of brachial plexus palsy. Injury at C5 and C6 is called Erb’s palsy, sometimes called Erb–Duchenne palsy. This is the most common level of involvement, present in approximately three-fourths of those with birth brachial plexus palsy (BBPP). Involvement of C8 and T1 is Klumpke’s palsy. It is debated whether Klumpke’s can occur in a birth brachial plexus injury, though it definitely occurs in other types of brachial plexus injury. The reason for this question is whether it is anatomically possible to have a C8 and T1 lesion alone without the involvement of C5 and C7. It appears that if there is an anatomic variation—for example, a rib, tendon, bony, or another anomaly that leads to the compromise of C8 and T1—this can occur in a birth brachial plexus injury. Otherwise, it appears that it cannot. Therefore, if a child presents with a C8 and T1 birth brachial plexus injury, it may be from anatomic anomaly, but there are two other options to consider. Most likely, it was initially a complete brachial plexus involvement but there was quick recovery of C5 and C7. This is likely, since the upper cervical root levels are relatively protected anatomically, so C8 and T1 may end up with the most severe injury. It is also possible that a spinal cord injury has been mistaken for brachial plexus palsy. All of these are important to consider during evaluation. There also may be complete brachial plexus palsy, including C5 and T1, with total motor and sensory loss. There also can be a variety of levels involved between upper plexus and total plexus palsy. EVALUATION Evaluation of patients with brachial plexus palsy includes clinical findings, electrodiagnosis, and MRI. There is debate about which of these is the most effective. MRI is expensive and requires sedation to perform on infants. It has been found to correlate with surgical findings 70% of the time, electromyography (EMG) 87% of the time, and clinical findings 60% of the time. The correlation was highest when all three of these were combined. MRI was effective only in those with C5, C6 root involvement (51). Clinical exam consists of a history and physical examination. The history includes the birth number of the child, the birth weight, and presence of maternal diabetes during the pregnancy, along with the size of previous infants and the birth size of the parents. The motor and sensory findings at birth, along with any change up to the time of evaluation, are important. The use of vacuum or forceps may be indicative of any difficulty with delivery. The most common association is shoulder dystocia. Other useful information is whether there were signs of bruising or other injuries, or whether there was involvement of the contralateral arm or the legs at delivery. Physical examination begins with visualization of the arm to include the size and bulk. A cool temperature may be noted in those with severe involvement. Sensory evaluation is critical to determine the extent and levels of involvement. Muscle stretch reflexes will be decreased or absent in the distribution of a brachial plexus injury. The primitive reflexes are also important. Since the upper plexus has more frequent involvement, the Moro reflex, which shows shoulder abduction and elbow flexion, is valuable in assessing those active movements. Torticollis is frequently seen, and usually this is with the face turned away from the involved arm. ROM is an important part of the evaluation since contractures are commonly seen in shoulder adduction and internal rotation, wrist flexion, forearm pronation, and even at the elbow into flexion commonly in later months and years. A key goal of the electrodiagnostic evaluation is to find subclinical nerve and muscle responses. The study must be specific depending on the clinical deficits noted, with studies performed that are pertinent to each individual’s examination. Sensory nerve conduction studies, motor nerve conduction studies, and EMG are performed. Diagnostic evaluation should include nontraditional nerve conduction studies, and frequently not the classic median and ulnar nerves, due to frequent involvement of only the upper brachial plexus. Axillary, musculocutaneous, and radial nerves are among those useful for electrodiagnostic study. Sensory nerve action potentials (SNAPs) are important, as these are most sensitive to axonal loss (52). The presence of SNAP responses in an insensate area is indicative of a preganglionic lesion, due to the location of the sensory cell bodies in the dorsal root ganglion. EMG may show activation of motor unit potentials in muscles with no clinical motor activity. Electromyographic evaluation is reported as being of some benefit, but it underestimates the severity of lesions (53). It has been recommended to be performed early in the first few days, then with a repeat evaluation after several months to more accurately identify cases where there is reinnervation occurring and therefore providing earlier determination of the need for surgical intervention (54). EMG at 1 month has been shown to have the best prediction for recovery in babies (55,56). Plain x-rays are useful in some clinical circumstances. Some abnormalities may mimic a brachial plexus palsy, including a fracture of the clavicle or humerus. Osteomyelitis may also mimic this, and has actually been reported as inciting temporary brachial plexus palsy (57). Neurofibromatosis or other tumors may also damage the brachial plexus. TREATMENT Education is initiated when a family is first seen. Therapy should be started as soon as possible after diagnosis. Positioning instruction begins immediately, and ROM exercises are generally initiated after 2 weeks. The wait is due to the fact that there is commonly noted pain with changing position of the shoulder for bathing or dressing in the first 2 weeks, so it appears that there is some early, short-term tenderness after the initial brachial plexus injury. It is also important to position the arm so that the baby will have maximal awareness of it. One way to accomplish this is with the use of a wrist rattle on the affected arm so that the baby’s attention can be drawn to that arm by sound or vision, because the weakness of that arm usually limits it from being moved in front of the face spontaneously. It is also recommended to have the family replicate movements with the affected arm that the baby spontaneously does with the unaffected arm, such as bringing the hand to the mouth. It is important that the family be taught to perform the exercise program several times a day. It is also important not to have such aggressive ROM in shoulder abduction or forearm supination that there is dislocation of the humeral head or radial head, respectively. Splinting is also commonly done by occupational therapy or physical therapy. Initially, there is frequently wrist drop, so splints may be made to provide optimal position of the wrist and fingers. Later on frequently there is an elbow contracture, so splinting is done to minimize that. Therapists also may do taping to help promote optimal positioning of the arm, particularly at the shoulder. Electrical stimulation is sometimes done for brachial plexus palsy, though this is frequently not tolerated at a very young age. Over time it does become accepted by many young children. Most commonly, it is performed with surface electrodes to increase muscle bulk by use of sufficient stimulation to get a local muscle twitch for approximately 20 minutes twice daily. It has been shown that continuous electrical stimulation to denervated muscles with implantable electrodes will lead to improved muscle outcome after nerve regeneration (58). Implantable electrodes have not been widely utilized and are not currently available on the U.S. market. It has been proposed that the adverse effects of prolonged denervation leave intramuscular axons deteriorated to such low numbers such that even with successful nerve regeneration, it is impossible to reinnervate enough muscle fibers for sufficient force (59). There are also proposals that low doses of brain-derived neurotrophic factor (BDNF) may protect against this decrease in those who have late nerve grafts, though high doses are inhibitory (60). COMPLICATIONS It is important to monitor for secondary complications. These commonly include muscle atrophy and joint contractures. Serial casting has been shown to decrease elbow flexion contractures (61). The affected arm frequently is shorter and has decreased circumference compared to the contralateral side. Joints may become dislocated, most commonly at the shoulder. Botulinum toxin injections decrease shoulder subluxation and decrease the need for surgery (62). Scapular winging is frequently seen. There may be torticollis, most commonly with the face turning away from the involved arm. General child development may be affected, including lack of awareness of the arm. Similarly, body image may be affected. There can be ulcerations from relatively minor trauma, particularly in insensate areas. Pain is infrequent after BBPP but common after brachial plexus palsy occurring later in life. Scoliosis has sometimes been linked to BBPP but two studies examining this question have found no correlation (61,63). SURGICAL INDICATIONS Indications for timing of brachial plexus surgery for infants have been controversial. It has been shown that a longer time for recovery leads to a worse shoulder function and that those who regain elbow flexion after 6 months of age have worse function than those who regain it between 3 and 6 months (64). Those with recovery by 3 months have normal function. Those who had microsurgery at 6 months did better than those who spontaneously recovered elbow flexion at 5 months (65). Surgical intervention is commonly recommended for those having less-than-antigravity strength in elbow flexion at 6 months of age (66). Estimates vary that from 4% to 34% of those with BBPP will require surgery for clinical improvement (67). Later brachial plexus injuries are divided into supraclavicular and infraclavicular injuries, supraclavicular being 75% and infraclavicular 25%. Supraclavicular injuries are generally felt to be due to traction of the plexus (classically in a motorcycle crash), and these have a worse prognosis than infraclavicular injuries (68). There may be a fracture of the clavicle or cervical transverse process, and supraclavicular fossa swelling may be seen. Dorsal scapular nerve or long thoracic nerve injury may be present. Supraclavicular lesions may also be due to falls; large objects falling on a shoulder, such as a tree limb; skiing or climbing; or contact sports, including football (52). Other etiologies are backpacks that are too heavy, tumors and gunshot wounds, or lacerations or animal bites. Those who have ipsilateral Horner’s syndrome and persistent pain have a worse prognosis (52). Infraclavicular brachial plexus injuries are more commonly associated with fractures and dislocations about the shoulder or humerus, occurring more often in older adults. The posterior cord, axillary nerve, or musculocutaneous nerve are classically involved. Infraclavicular injuries are less severe and have better outcomes (69). Infraclavicular plexus injuries may also be due to falls, motor vehicle collision, or tumors (52). Gunshot wounds, stab wounds, and failed attempts at shoulder reductions may cause infraclavicular injuries as well (70). Brachial plexus palsy has been reported after axillary crutch use, anesthesia positioning (particularly with table tilt), and after bony fracture with malunion (71). For severe injuries later in life, recommendations are for surgical exploration and nerve grafting, most commonly at 3 to 4 months postinjury (70,72). SURGERY Surgical interventions for brachial plexus palsy are varied. There may be electrical testing, including evoked potentials, and nerve conduction studies done to assess the nerves in the operating room to be as specific as possible with the procedures undertaken. Microsurgical repair yields results months later. Recovery is generally felt to proceed at the rate of approximately a millimeter a day or an inch a month. It is also believed that there is more nerve growth factor available in babies than older people so that both size and age have an impact in outcome. It is critical to have therapy postsurgery and to continue a faithful daily home program as well. There are a variety of options for surgical procedures for brachial plexus injury. Neurosurgery may include neurolysis in which scar and fibrotic tissue are removed from nerve tissue. Direct nerve transfers have the advantage of quick recovery time due to short regeneration distance versus neurotization, which requires interposition of a nerve graft. The sural nerve and great auricular nerve are commonly used as donor nerve fibers for these grafts (73). More recently, end-to-side neurorrhaphy is performed for those who have some intact fibers for augmentation. The advantage of this is not requiring a sacrifice of any other nerves. Not uncommonly, synkinesis of newly innervated muscles with contraction of muscles innervated by the donor nerve may be seen, and is treated with therapy (74). Synthetic nerve conduits are now available for nerve grafting. Some classic nerve procedures involve transfer from a functionally less important nerve to a distal denervated nerve. Common examples include taking intercostal nerves to the upper trunk or to the suprascapular nerve. Another classic surgery is the Oberlin procedure, which transfers one or several ulnar nerve fascicles to the musculocutaneous nerve as it enters the biceps muscle (75). Transfer of the spinal accessory nerve to the suprascapular nerve is also commonly used for shoulder abduction. Contralateral C7 transfers have been performed both in adults and infants for those with multiple severe avulsions. This procedure has been shown to provide adequate elbow flexion as a result, and most patients have had only temporary sensory deficits on the ipsilateral C7 side (76). This procedure clearly illustrates the point that nerve grafts are not required to have their original source but can have function coming from a variety of intact neurologic structures. This allows for greater flexibility and creativity in the surgeon performing the procedure, aiming for recovery of function. Glenoid dysplasia with posterior shoulder subluxation is frequently a complication of children after BBPP. It was commonly thought to be the result of a slowly progressive glenohumeral deformation due to muscle imbalance and possible physeal trauma, but it was found that posterior shoulder dislocation happened at a mean age of 6 months, with rapid loss of passive external rotation. There was no correlation between the initial neurologic deficit and the presence or absence of dislocation (77). Many musculotendinous surgical procedures are performed for children with BBPP. It has been shown that latissimus dorsi and teres major tendon transfer to the rotator cuff, along with musculotendinous lengthening, will provide improved shoulder function but no significant change in the bony position of the shoulder or humerus. This procedure does not decrease glenohumeral dysplasia (78). With internal rotational contracture and glenohumeral joint deformity, along with significant abnormality of glenohumeral joint, a derotational osteotomy can result in improved shoulder function, along with improved internal rotation contracture (79). Some children with BBPP have been described to have arthroscopic release of shoulder deformity alone before 3 years, and for those over 3 years of age, arthroscopic release with latissimus dorsi transfer. They all show improved shoulder position, but they do have loss of internal rotation. Some of the children under 3 years of age do have a recurrence and require a second procedure with a latissimus dorsi transfer (80). In adults, performing a glenohumeral arthrodesis, in patients with upper plexus palsy with functional distal arm, as well as in those with total plexus palsy, has been shown to increase functional capabilities. The strength of the pectoralis major is a significant prognostic factor for outcome (81). Performing wrist arthrodesis in adults with brachial plexus injury is done for improved function as well as pain relief. There will be limitations after having this procedure, and potential patients need to have full information in order to know what to expect prior to the procedure. There also remains some controversy regarding the ideal position to place the hand, which is generally placed in slight wrist extension and ulnar deviation in order to have the most powerful grip (71,82). A dramatic surgical procedure sometimes performed for children and adults with brachial plexus palsy is a free muscle transfer, most commonly performed with the gracilis muscle. The muscle is transferred with its vascular and nerve supply and attached to these in the arm. This procedure has been described as having reliable results for elbow flexion and wrist extension (71). PAIN Pain has not been reported as a severe problem in birth brachial plexus injury, although with one study reporting biting of the limbs in less than 5% of the cases, it is possible that this is a manifestation of pain. Self-mutilation has been reported in youngsters after a birth brachial plexus injury. A study of 280 patients with a birth brachial plexus injury found that 11 of these children had self-mutilating behavior by biting or mouthing the affected arm. The age of onset was between 11 and 21 months, and the duration of the behavior was 4 to 7 months. This was more frequent in children who underwent surgery, with 6.8% of these children, and 1.4% of children who did not have surgery. It is unclear if this is due to surgery or the severity of the injury or a combination of these (83). It is also possible that this is a response to the unusual sensation of the recovering nerve, possibly a manifestation of what we see on examination as a Tinel’s sign. It has been felt, however, that it is more likely biting with the resumption of nerve growth with sensation of tingling as there is recovery occurring, but this is not proven. In those who have later traumatic or nontraumatic brachial plexus injuries, pain can be a significant problem. It has been described most commonly with avulsions as severe burning and crushing pain most commonly in the hand. This may develop days to months after the injury and almost always within 3 months. It is most commonly resolved within several years, but approximately 20% of those with pain have severe, long-lasting disruptive pain (84). This can be treated with transcutaneous nerve stimulation classically from C3-T2. Medications, including antidepressants and anticonvulsant agents, have been affective. Topical treatments, including topical lidocaine 5% pain patches, are sometimes useful. Nerve surgery is commonly effective in resolving pain (85,86). The author has seen children with traumatic brachial plexus injuries and severe pain complaints prior to their nerve procedure wake up postoperatively in the recovery room excited that the pain is gone. Amputation is not effective for resolving the pain (87). REHABILITATION OF THE CHILD WITH RHEUMATIC DISEASE Rehabilitation of the child with rheumatic disease requires an interdisciplinary approach that includes the child and family. Although most often the physiatrist is not the treating physician in rheumatologic disease, they can play a key role in the comprehensive management of these conditions, along with other members of the rehabilitation team, to maintain or restore age-appropriate function and development, prevent deformity and contractures, and help manage pain. JUVENILE IDIOPATHIC ARTHRITIS Juvenile idiopathic arthritis (JIA), formerly known as juvenile rheumatic arthritis (88), is the most common rheumatic disease of childhood, affecting approximately 16 to 150 in 100,000 (89). In 1995, the International League Against Rheumatism (ILAR), together with the World Health Organization, reclassified chronic childhood arthritis (90); the second revision occurred in 2001 (91). Chronic childhood arthritis is now known as JIA and is divided into the following seven subtypes: systemic arthritis, oligoarthritis, rheumatic factor (RF)-negative polyarthritis, RF-positive arthritis, psoriatic arthritis, enthesitis-related arthritis, and undifferentiated arthritis. JIA occurs in children before the age of 16 years, persists at least 6 weeks, and has had other known conditions excluded; etiology is unknown, but seems to include genetic and environmental components (89,92). In 2011, the American College of Rheumatology (ACR) published recommendations for the treatment of JIA, organized into five treatment groups; namely, arthritis of four or fewer joints, arthritis of five or more joints, active sacroiliac arthritis, systemic arthritis with active systemic features (and without active arthritis), and systemic arthritis with active arthritis (and without active systemic features) (93). In October 2013, these recommendations were further reviewed and updated, with emphasis on systemic JIA (94). Both recommendations will be discussed further in the treatment section. Early arthritis may be manifested by swelling, warmth, and joint stiffness, typically worse at the beginning of the day then improving with activity. Symptoms usually fluctuate; uncontrolled inflammation leads to joint damage. Younger children rarely complain of joint pain, but may instead become irritable, stop walking or using an extremity, or regress in their behavior (95). Other symptoms include decreased appetite, malaise, inactivity, morning stiffness, nighttime joint pains, and failure to thrive (95). Enuresis may occur in a recently toilet-trained child (96). Later disease presents with reduced ROM, contractures, overgrowth or undergrowth of affected limbs, and resultant disability. A characteristic feature of chronic arthritis in children is the effect the disease has on bone and joint development (97,98). Local growth disturbances at inflammation sites can lead to overgrowth (secondary to possible inflammatory-mediated increased vascularization and growth factor release) or undergrowth (secondary to growth center damage or premature fusion of epiphyseal plates). Irregular traction on growing structures secondary to muscle spasms and periarticular fibrosis can also cause aberrant growth (97,98). Micrognathia, leg-length inequalities, and developmental hip anomalies are all possible results from these processes. Steroids can also contribute to severe growth effects, as well as osteoporosis (99). The differential diagnosis of JIA is large (Table 10.1 provides a full differential diagnosis). The assumption that JIA will universally resolve by adulthood is incorrect (100). Radiological joint damage occurs in children with systemic arthritis and polyarticular arthritis within 2 years, and in oligoarthritis within 5 years (101,102). Despite long-term persistence of disease activity in JIA, much improvement in functional outcomes has been made in the past decade (103,104). Indicators of poor outcome include greater severity or extension of arthritis at onset, symmetrical disease, early wrist or hip involvement, presence of RF, persistent active disease, and early radiographic changes (105). In the 2011 ACR recommendations, features of poor prognosis are further specified by treatment group. Features of poor prognosis for three of the five categories, arthritis of four or fewer joints, arthritis in five or more joints, and systemic arthritis with active arthritis (and without systemic features), include arthritis of the hip or cervical spine. Four of the five treatment groups (all except for systemic arthritis with active systemic features [and without active arthritis]) show poor prognosis with radiographic damage (erosions or joint space narrowing by radiograph) (93). TABLE 10.1 DIFFERENTIAL DIAGNOSIS OF JUVENILE IDIOPATHIC ARTHRITIS CLINICAL FEATURES OF JIA SUBTYPES Systemic JIA Systemic-onset JIA presents with many extra-articular features and represents 10% to 20% of all JIA (92). Diagnosis requires arthritis in more than one joint for at least 6 weeks’ duration in a child aged below 16 years accompanied or preceded by quotidian fever (spikes >39 degree Celsius once a day with return to normal between peaks) of 3 days’ duration, plus one or more of the following: evanescent salmon-colored rash, generalized lymphadenopathy, hepatomegaly, splenomegaly, or serositis (91,94). The goal of treatment for systemic JIA is similar to other categories of JIA, and focuses on the prompt control of active inflammation and symptoms, and the prevention of disease- and/or treatment- related morbidities such as growth disturbances, joint damage, and functional limitations (106). About 5% to 8% of children with systemic JIA develop a life-threatening complication known as Macrophage Activation Syndrome (107) with persistent fever, lymphadenopathy, and splenomegaly, and there is profound depression in one or more of the blood cell lines (often initially platelets) with raised liver function enzymes and clotting abnormalities. Definitive bone marrow examination shows numerous well-differentiated macrophages actively phagocytizing hematopoietic elements (108). In one-half of children with systemic JIA, the course follows a relapsing–remitting course, with arthritis accompanying febrile episodes, then remission once systemic features are controlled. Long-term outlook for these children is usually good. In the other half, the disease is unremitting, with resultant severe joint destruction, and is probably the most severe JIA subtype (89,109). Poor prognostic signs include the continued presence of systemic features and a platelet count exceeding 600,000/mm3, 6 months after onset (95). At least one-third of children will develop severe arthritis (110). Oligoarthritis Oligoarthritis is classified into two subtypes: persistent (affecting not more than four joints throughout the disease course) and extended (affecting more than four joints after the first 6 months of disease). Characteristically, there is an early onset before 6 years of age of an asymmetric arthritis, usually in the lower limbs, and predominantly in females. Antinuclear antibodies (ANAs) are detected in substantial titers in about 70% to 80%, and they represent a risk factor for iridocyclitis. Children with the oligoarthritis subtype generally have the best outcome (89); however, sight-threatening, clinically silent uveitis develops in the first 4 years from diagnosis. Regular ophthalmology follow-up is essential (111). Polyarthritis Polyarthritis must affect five or more joints in the first 6 months of the disease. RF-positive polyarthritis mainly affects adolescent girls, with a symmetrical pattern, and is the same as adult RF-positive disease (97). By 5 years from onset, severe deforming arthritis is generally present (98). RF-negative polyarthritis is a more heterogeneous group with more variable outcome. Approximately 20% to 40% of those affected are ANA-positive, and chronic uveitis is found in 5% to 20% (97); it is believed by some authors that this entity represents a later stage of early-onset oligoarthritis (112). Future versions of the ILAR classification of JIA may explore this more fully. Psoriatic Arthritis Psoriatic arthritis accounts for about 5% of JIA and requires the simultaneous presence of arthritis and the typical psoriatic rash, or if the rash is absent, arthritis plus two of the following: positive family history of psoriasis in a first-degree relative, dactylitis, and nail pitting. Psoriatic disease in children before the age of 5 years appears to be more difficult to control than in an older subset of children, with a median of 9.5 years (92). Enthesitis-Related Arthritis Enthesitis-related arthritis affects males after the age of 6 years (97,98) and most children are human leukocyte antigen (HLA)-B27-positive. The most common sites of enthesitis are the calcaneal insertion of the Achilles tendon, plantar fascia, and tarsal area. Arthritis commonly affects the joints of the lower extremities. Unlike other JIA subsets, hip involvement is common at disease presentation. These children may progress to fulfill criteria for ankylosing spondylitis, reactive arthritis, or arthritis associated with inflammatory bowel disease. Uveitis is also a clinical problem in this subset, but it is usually sudden in onset, symptomatic, and more unilateral than in children with other JIA subsets (92). Juvenile ankylosing spondylitis, not considered part of the JIA subclassification; mainly affects adolescent boys; is strongly associated with HLA-B27; and manifests as an asymmetric, often episodic, oligoarthritis in the lower limbs. Later on, bilateral sacroiliac joints become involved, and progression of the disease can lead to the characteristic “bamboo” spine on radiographic images secondary to ankylosis of spinal joints. In children, peripheral arthritis and enthesitis present early in the disease, but sacroiliac and spine joints are not involved until many years later (113). Rehabilitation involves maintaining spinal ROM through extension exercises, strengthening hip extensors and quadriceps muscles, custom shoe inserts to relieve pain, and deep breathing exercises to maximize chest expansion. Because of the chronic course of the disease, the child and parents should not restrict age-appropriate social and recreational activities (113). Inflammatory bowel associated arthritis occurs in approximately 10% to 20% of children with ulcerative colitis and Crohn’s disease. The arthritis usually affects a few joints and may be associated with spondylitis; erythema nodosum and growth failure may occur. Undifferentiated Arthritis This subset is not a separate entity, but is more of a catchall category for those children who do not satisfy inclusion criteria for any category, or who meet criteria in more than one category. REHABILITATION OF THE CHILD WITH JIA Goals of treatment include controlling symptoms, preventing joint damage, achieving normal growth and development, and maintaining function and normal activity levels. Treatment goals may vary during maintenance and acute flare-ups of the disease. Resting a joint may be necessary during an acute flare-up to prevent aggravation of the disease process; activities that affect or excessively stress joints should be discouraged during acute flare-ups. Resting a joint may also be useful during the maintenance phase for joint protection. Rest periods may be necessary to reduce fatigue; resting in the prone position will help reduce hip and knee flexion contractures. Splinting is used during a flare-up to provide alignment during a rest period. Functional splints may be used during flare-ups and maintenance phases if they provide joint relief and allow functional activities without stressing inflamed joints. Splinting can be used during the maintenance phase to promote local joint rest, support weakened structures, and assist function. To prevent flexion contractures, the upper extremity is splinted in a functional position as follows: wrist 15 to 20 degrees of extension, some finger flexion, 25 degrees at the metacarpophalangeal (MCP) joint, and 5 to 10 degrees at the PIP joint, with the thumb in opposition. Ring splints can be used for finger deformities. Knee immobilizers may be used to maintain knee extension at night; rotate on alternate legs for better compliance. Dynamic splints or serial casts can increase ROM. Foot orthoses can promote arch support and reduce pain in weight-bearing. Gentle ROM with passive extension greater than flexion two to three times a day is used to preserve joint ROM. Incorporating pain medication, progressive muscle relaxation, breathing exercises, biofeedback, massage, or doing the exercises in a nice, warm tub can greatly facilitate ROM exercises. Gentle ROM exercises should be done as tolerated during acute flare-ups to prevent flexion contractures. Heat is an excellent modality in the maintenance phase to decrease stiffness, increase tissue elasticity, and decrease pain and muscle spasm. Hydrotherapy with temperatures 90 to 100 degrees Fahrenheit, fluidotherapy, paraffin, or moist heat can be used. Most children prefer heat to cold. Taking a hot bath or shower, sleeping in a sleeping bag, or using a hot pack (along with ROM exercises) may help relieve morning stiffness. Caution must be exercised in insensate areas to avoid burns. Ultrasound is contraindicated in children with open growth plates. Heat should not be used during an acute flare-up, as it increases the inflammatory response and causes further joint destruction. Cold can be used during an acute flare-up for pain relief and to decrease swelling. It may also be beneficial during the maintenance phase for the same reasons. Cold should not be used over insensate areas or in those with Raynaud’s phenomenon. Adaptive strengthening exercises can be incorporated into play and recreational activities. Some examples include throwing a ball (strengthens elbow and shoulder), riding a bike (promotes knee and hip extension), and swimming (decreases weight-bearing on painful joints). Incorporating general aerobic conditioning is also important and may include activities such as swimming, dancing, noncontact karate, and tai chi. Isometric strengthening exercises are fine during an acute flare-up, but vigorous exercise should be avoided until the acute process is over. Hydrotherapy can be combined with land-based physiotherapy in treating JIA (114). Adaptive equipment can be used for joint protection, rest, and to minimize further joint destruction during both phases. Examples include adaptive utensils, adaptive pens and computer access, table and desk modifications (to prevent excessive trunk and neck flexion), zipper pulls, dressing sticks, long-handled brushes, elastic waistbands, Velcro closures, and larger buttons. Children should actively participate in functional activities of daily living (ADLs) training in order to choose acceptable devices and improve their use. Activity and ambulation should be encouraged as much as possible. A posterior walker for upright posture (with decreased flexion) and a standing program may be useful for functional mobility training if wheelchair use cannot be avoided. In children with JIA, custom-made semirigid foot orthotics with shock-absorbing posts have been found to significantly improve pain, ambulation speed, self-rated activity, and functional ability levels compared to prefabricated off-the-shelf shoe inserts or supportive athletic shoes alone (115). A presurgical joint rehabilitation program aims to strengthen the muscles needed for mobility in the postoperative period, train for future ambulation aids, and identify other joint involvement that may affect the rehabilitation process. Postsurgical rehabilitation fulfills those goals set in the presurgical rehabilitation program. Ambulation aids such as the platform walker may be used to better distribute weight-bearing pressure on affected upper extremity joints after knee or hip surgery. In children, post hip prosthesis, the acetabular component should be checked for loosening (as opposed to the femoral component in adults), especially if children are active. Growth retardation can occur during periods of active disease; it may also be compounded by corticosteroid use. Maximize growth by promoting optimal nutrition. Children with JIA should eat a balanced diet with supplemental multivitamins, calcium, vitamin D, and sunshine secondary to the high risk of osteopenia. Plenty of (nonimpact) activity again should be encouraged. Counseling for both the child with JIA and his or her family should be provided to maximize psychosocial and emotional well-being. Treatment goals also include addressing family, school, and vocation. Assisting in the preparation of a 504 plan for school accommodations enables a child with joint disease opportunity for more complete participation in his or her school life and academic career. Summer camps are a practical way of addressing peer support within adolescent rheumatology services; positive effects include increased control, self-esteem, physical fitness, independence from parents, self-management of health care, and an opportunity to meet others with a similar condition (116). SPECIFIC JOINTS IN JIA Cervical Spine Cervical spine involvement occurs more often in children with JIA than adults. Restriction of ROM, pain, and muscle spasms, which may present as torticollis, may be seen. A soft cervical collar to serve as a reminder for proper alignment and provide warmth may be helpful in acute pain with muscle spasm. Minimizing time in flexion is important. If the transverse ligament becomes weakened, atlantoaxial subluxation can occur. If subluxation occurs, a firm cervical collar should be worn during automotive transport. Temporomandibular Joint (TMJ) This joint is affected in almost two-thirds of children with JIA (117) by causing pain in chewing and opening the mouth, stiffness, and micrognathia. Younger children will not complain of jaw pain, but will instead choose to modify their diet to avoid pain. Progressive jaw ROM exercises and modalities may help treat pain and stiffness. If the lower jaw does not develop properly, it may create an overbite, requiring orthodontist intervention and/or oral surgery. Mandibular and facial growth disturbances are more common in polyarticular types of JIA. Upper Extremities The shoulder is not commonly involved at the onset of disease. Approximately one-third of children with polyarticular or psoriatic disease may eventually develop shoulder involvement and loss of adduction and internal rotation affecting midline ADLs, such as grooming and toileting. The elbow requires at least 90 degrees of flexion range to perform ADLs such as eating, grooming, and reaching. Loss of more than 45 degrees of elbow extension limits the use of arms as levers to rise from a seated position and makes toileting and lower extremity dressing difficult. Wrist involvement is common in children; there is early loss of wrist extension with progressive flexion contracture. A nighttime resting wrist splint can maintain the wrist in 15 to 20 degrees of extension with the fingers in a few degrees of flexion; ulnar deviation can also be built in as necessary. Strengthening of wrist extensors and radial deviators is necessary to reduce wrist flexion and ulnar deviation contractures. Moist heat to reduce spasm and improve tissue elasticity followed by serial casting for 48 to 72 hours as tolerated may help reduce contractures by slowly increasing wrist extension while controlling ulnar deviation and subluxation; commercially available dynamic splinting may also facilitate stretching. Should ankylosis be inevitable, the hand should be splinted in a neutral position for optimal function in self-care. Functional grasp may become limited as fingers lose both flexion and extension range. Flexion contractures of the metacarpal and PIP joints are often seen. The use of ring splints in metal or plastic can help control PIP flexion and extension seen in boutonniere and swan neck deformities, respectively. Fingers can be strengthened through play with clay and various adaptive putties. Lower Extremities In the lower extremities, flexion contractures occur at the knee and hip. Painful ambulation can lead to increased sitting, which in turn leads to increased flexion contracture, deconditioning, weakness, atrophy, and osteoporosis. Hip flexion contractures in children occur with internal rotation and adduction, compared with adults who tend to develop external rotation and abduction. Prone lying greater than 20 minutes per day with the hips and knees extended and feet off the edge of the bed can help prevent these contractures. Other strategies include strengthening of the hip extensors, external rotators, abductors, and quadriceps, along with ROM exercises to stretch the hip flexors, internal rotators, adductors, and hamstrings. Hip extensors can be strengthened through swimming, aquatic therapy, and bicycling. Encouraging upright posture and ambulation, using a stander as necessary, is also helpful. Hip development may be assisted by the use of a stander; a prone stander can strengthen neck and hip extensors, while a supine stander maintains the knees in extension and allows upright weight-bearing. The knee is the most commonly affected joint in JIA; early involvement of the knee can cause quadriceps weakness that may not resolve. Knee contractures can lead to other joint contractures and further gait abnormalities. Bony overgrowth with resultant leg-length discrepancies (LLDs) is often seen. The knee can be maintained in extension using resting splints such as knee immobilizers and alternating legs every night as needed to increase comfort and compliance. Dynamic splinting using an adjustable knee joint can be used to improve ROM and limit excessive flexion and valgus tendency. Because forced extension of the knee with a contracture can exacerbate posterior subluxation, caution must be exercised in using bracing and splinting. Active quadriceps strengthening should be done post brace removal and also maintained with knee extension exercise or isometric exercises if too painful. Kicking, bicycling, and walking can also strengthen weak quadriceps muscles. Multiple foot deformities can occur in JIA, including claw toe, valgus or varus hindfoot, and ankle plantarflexion contracture deformities. The mid-foot is frequently affected, and can be quite painful and difficult to treat. Tenosynovitis that is difficult to discern from joint disease may occur. Molded foot orthoses can be used to reduce pain at the metatarsal heads and heels with weight-bearing. A University of California at Berkeley orthosis can prevent or control varus and valgus deformities. A posterior leaf-spring ankle foot orthosis (AFO) or nighttime resting splint may be helpful to reduce loss of ankle dorsiflexion range and control varus and valgus. Ankle rotation exercises, balancing exercises, and raising the heel on a step can strengthen ankle muscles. Footwear should be comfortable and accommodate any foot deformities. High heels should generally be avoided, as they can help develop plantarflexion contractures and add to foot deformities. Flip-flops should also be avoided secondary to their lack of adequate support. Inflammation causing bony overgrowth at the distal femur can cause a true LLD, leading to pelvic asymmetry and scoliosis. The increased blood flow from inflammation may alternatively cause early epiphyseal closure and overall limb shortening. MEDICAL AND SURGICAL TREATMENTS OF JIA Children with JIA are treated with more of an induction and maintenance approach, taking advantage of windows of opportunity to modify the disease course, usually under the guidance of a pediatric rheumatologist (118). Treatment is aimed at inducing remission with the least medication toxicity. In 2011 and 2013, the ACR published recommendations for the treatment of JIA organized into five treatment groups as previously discussed (93,94). Within the five treatment groups, treatment is stratified into three disease activity levels (low, moderate, and high). Choice of treatment is guided by disease severity and the presence or absence of poor prognostic features. In the first treatment group, history of arthritis in four or fewer joints, escalation of therapy typically proceeds from nonsteroidal anti-inflammatory drugs (NSAIDs) to intra-articular glucocorticoid injections to methotrexate to TNF-α inhibitors. In children with low disease activity (single joint, normal inflammatory markers, etc.), monotherapy with NSAIDs may be sufficient. Intra-articular injections of triamcinolone should provide relief for at least 4 months, and can be repeated if helpful. Methotrexate should be started as initial treatment in children with high disease activity and features of poor prognosis, or in those with lower disease activity who fail to show adequate response with NSAIDS and triamcinolone injections. In enthesitis-related JIA, sulfasalazine rather than methotrexate is recommended at a similar stage. After 3 to 6 months (depending on disease activity) of methotrexate with inadequate response, TNF-α inhibitor treatment should be considered. In children with a history of arthritis in five or more joints (does not require five currently active joints), treatment with initial NSAIDs is more quickly escalated to methotrexate, with joint injection as needed. In moderate disease activity and poor prognostic features, or in children with high disease activity, treatment may start with methotrexate. Leflunomide is sometimes used as an alternative to methotrexate. The IL-6 inhibitor, tocilizumab, is now FDA-approved in children above 2 years of age and older with active polyarticular JIA (119). Tocilizumab can be used alone or in combination with methotrexate (120). Escalation to a TNF-α inhibitor follows in 3 to 6 months dependent on disease characteristics and severity if there is inadequate response to methotrexate or leflunomide. If there is still no response with a TNF-α inhibitor after 3 to 4 months, another TNF-α inhibitor can be tried, or abatacept (a T cell inhibitor). Rituximab may be used for nonresponders to the previously mentioned regimen in those children with RF-positive polyarticular JIA. In children with the third treatment group, active sacroiliac arthritis, use of a TNF-α inhibitor is recommended more readily than in any other treatment group. A TNF-α inhibitor may be started after failure of an adequate trial of NSAIDs or methotrexate or sulfasalazine for 3 to 6 months (depending on disease characteristics and severity). In the 2013 recommendations, systemic JIA was broken down into three phenotypes: significant systemic features and varying degrees of synovitis, significant arthritis and no significant systemic features, and features concerning MAS (94,121). In children with significant systemic features and varying degrees of synovitis, initial treatment with anakinra (an interleukin 1 receptor inhibitor) or corticosteroids was recommended in most cases. For children without systemic features and with varying degrees of active synovitis, initial treatment recommendations were methotrexate or leflunomide for an active joint count higher than 4, with change to abatacept, anakinra, a tumor necrosis factor α inhibitor, or tocilizumab (120,106) if there is inadequate response. The interleukin 1 β inhibitor, canakinumab, has also been FDA-approved for systemic JIA (122). NSAIDS or intra-articular triamcinolone joint injections were recommended as initial treatment for children with four or less active joints (106). For children with MAS, which carries an estimated 6% or more mortality rate, initial treatment recommendations include anakinra, a calcineurin inhibitor, or systemic glucocorticoid monotherapy for up to 2 weeks. The use of abatacept, intravenous immunoglobulin, or tumor necrosis factor α inhibitors is inappropriate for children with MAS (94,121). It is stressed that systemic JIA is a very different disease from polyarticular JIA and that children need to be treated aggressively with biologics right away. Moving up to the next step after 1 month of inadequate response is recommended (121). The reader is encouraged to check the ACR website, www.rheumatology.org, for the most current, detailed recommendations for treatment of JIA. Early and aggressive treatment of JIA with newer agents holds unlimited promise for even better outcomes for children with JIA. Steroids are used as sparingly as possible to control inflammation in order to avoid long-term side effects such as weight gain, poor growth, and risk of infection. There is no systemic evidence that steroids are disease-modifying (100). Children with JIA are at high risk of developing osteopenia secondary to the disease itself, to steroid treatment of the primary disease, lack of physical activity and weight-bearing, limited sunshine exposure, and inadequate vitamin D and calcium. Calcium and vitamin D supplementation, sunshine, and encouragement of physical activity should be incorporated into the treatment plan. INFECTIOUS DISEASE WITH ARTHRITIS Infectious causes of arthritis include bacterial, viral or postviral, and fungal. Osteomyelitis and reactive arthritis can also be confused with JIA. SEPTIC ARTHRITIS Joint involvement in septic arthritis may be by hematogenous spread, direct extension from local tissues, or as a reactive arthritis. Bacterial septic arthritis is usually monoarticular in children, but multiple joints can be involved. Children may present with fever, joint pain, and decreased joint mobility, especially in the knees, hips, ankles, and elbows. A child may not allow the affected joint to be touched and, sometimes, may not even allow the affected joint to be seen. An ambulatory child will refuse to bear weight on the affected extremity. Premature infants presenting with irritability, fever, and hips positioned in abduction, flexion, and external rotation should be checked for septic arthritis of the hip. Boys 3 to 10 years who present with hip or referred knee pain should be checked for transient synovitis. Ear infections are the most common source of bacteria leading to septic arthritis in children (123). Osteomyelitis or diskitis can develop in children with septic or reactive arthritis. In all age groups, 80% of cases are caused by gram-positive aerobes (60% S. aureus; 15% beta-hemolytic streptococci; 5% Streptococcus pneumoniae), and approximately 20% of cases are caused by gram-negative anaerobes. In neonates and infants younger than 6 months, S. aureus and gram-negative anaerobes comprise the majority of infections. Clinically affected joints require emergent aspiration and treatment. Aspiration of joint fluid is necessary for possibly identifying the agent and relieving pain. Joint fluid reveals increased white blood cells (WBCs), protein, and low-to-normal glucose. Radiographic findings progress from soft tissue swelling to juxta-articular osteoporosis, joint space narrowing, and erosion. Treatment consists of appropriate antibiotic therapy, joint aspiration to relieve pressure and pain, and physical therapy to maintain ROM. REACTIVE ARTHRITIS Reactive arthritis is different from septic arthritis in that it is an autoimmune response triggered by antigen deposit in the joint spaces; synovial fluid cultures are negative. It is set off by a preceding infection, the most common of which would be a genital infection with Chlamydia trachomatis in the United States, usually in adult males (124). Reactive arthritis after Yersinia and Campylobacter can be associated with HLA-B27. Yersinia enterocolitica infection can show persistence of the organism in joint fluid, especially the knee. The main goal of treatment is to identify and eradicate the underlying infectious source with appropriate antibiotics, if still present. Analgesics, steroids, and immunosuppressants may be needed for patients with severe reactive symptoms that do not respond to any other treatment. LYME DISEASE Lyme disease is caused by the spirochete, Borrelia burgdorferi, with transmission to humans via the deer tick, Ixodes dammini. Lyme disease is the most common tick-borne disease in North America and Europe. The initial phase of Lyme disease (lasting about 4 weeks) consists of fever, fatigue, headache, arthralgias, myalgias, stiff neck, and erythema migrans. Erythema migrans looks like a reverse target skin lesion, as it is a large, red lesion with a central clearing area; it occurs 1 to 30 days after the tick bite. The late phase, lasting months to years, is characterized by arthritis, cardiac disease, and neurologic disease. Intermittent episodes of unilateral arthritis involve the knee most often; hip, shoulder, elbow, wrist, and ankle may also be involved. In 85% of children, the arthritis resolves before the end of the initial treatment; in 10%, a chronic inflammatory phase develops. OTHER RHEUMATIC DISEASES OF CHILDHOOD Systemic Lupus Erythematosus Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease with widespread immune complex deposition that results in episodic inflammation, vasculitis, and serositis. Children are more likely than adults to present with this systemic disease; 20% of cases begin in childhood. Females are affected 4.5 times more than males. One-third of children have the erythematous butterfly rash over the bridge of the nose and cheeks; this rash may occur after exposure to sunlight. Most children develop a transient, migratory arthritis of the extremities; radiographic evidence of joint deformity and erosion is not common. Pain may be out of proportion to joint findings on examination. Proximal muscle weakness may be a result of acute illness, myositis, or the result of steroid-induced myopathy. Long-term steroids also increase the risk of avascular necrosis of the femoral head. Systemic features of SLE may include pericarditis or endocarditis; proliferative glomerulonephritis or other renal disease; seizures, psychosis, memory deficits, headaches, or behavior changes; pulmonary hypertension and/or hypertension. Nephritis occurs in approximately 75% of children with SLE and is the main factor for determining outcome. Hematuria, proteinuria, persistent hypertension, chronic active disease, and biopsy proven diffuse proliferative glomerulonephritis are associated with a poor outcome. Ten-year survival is approximately 80%, although this number is lower in lower socioeconomic populations. Management of SLE is symptomatic. Maintaining physical activity as much as possible, avoiding excess sunlight exposure, optimizing nutrition, and providing adequate social supports are key. For some children with open discoid lupus rash lesions, dressing changes and wound cares may be best facilitated with individualized whirlpool therapy, much like is used for burn wound cares. NSAIDs are mainly used for arthritis and musculoskeletal conditions. Fever, dermatitis, arthritis, and serositis usually resolve quickly with low-dose steroids, whereas serologic findings may require weeks of steroid therapy. Hydroxychloroquine may be used for skin manifestations or in concert with steroids to lower the steroid dose. High-dose steroids, immunosuppressive agents, and biologic agents may be necessary for more severe disease manifestations. Scleroderma Systemic sclerosis is uncommon in children; linear and focal cutaneous involvement is most common in children. Girls between ages 8 and 10 years are more often affected; duration can last 7 to 9 years. Linear scleroderma presents with atrophic, erythematous skin areas, which later become fibrotic. This skin then binds to underlying subcutaneous tissues, and underlying muscle and bone also become involved. Children may have pain from these skin changes. Soft tissues can atrophy, leaving areas of asymmetry. Scleroderma en coup de sabre is a unilateral linear involvement of the face and scalp, often with loss of hair on the involved side, with loss of facial asymmetry. Systemic disease in children is uncommon. Physical therapy is necessary to prevent loss of ROM and contractures because of the cutaneous involvement. Soft tissue massage, moist heat, stretching, and ROM exercises help maximize joint mobility. Topical corticosteroids may be helpful in treating localized skin disease; systemic steroids, methotrexate, and physical therapy may alter the course of progressive disease. HEMATOLOGIC DISORDERS Hemophilia Hemophilia is a bleeding disorder that affects about 18,000 Americans; each year, about 400 babies are born with the disease, and it occurs in 1 out of every 7,500 males. Of these, about 85% of cases are Factor VIII (hemophilia A) and 14% are Factor IX (hemophilia B). In hemophilia, bleeding occurs without any recognizable trauma; spontaneous bleeding happens most often in the knees, ankles, elbows, and shoulders. Bleeding into the joints usually begins after a child begins to walk. As bleeding begins, the child may experience warmth or tingling in the joint. As bleeding progresses, there is usually a feeling of stiffness, fullness, and pain. The joint swells and may be warm and tender, causing synovial membrane thickening. Without treatment, hypertrophy of the synovium with its increased vascular supply, creates a cycle of more bleeding and destruction. Without intervention, fibrosis and arthritis set in, making joint replacement at an early age the only option. Pain and swelling can also lead to decreased active joint ROM, further leading to contractures. Other complications include muscle atrophy, osteopenia, peripheral neuropathy, and compartment syndrome. The main treatment for hemophilia is injections of cryoprecipitate. Acute hemarthrosis requires joint immobilization for 48 hours to prevent further bleeding. Once pain and swelling subside, passive ROM should be started to prevent fibrosis and contracture development. Analgesics, anti-inflammatory medications, and aspiration of blood from the joint if overlying skin is tense are important in pain management. Joint function may be regained in 12 to 24 hours with early factor replacement, but may take up to 2 weeks for more blood reabsorption (125). ROM exercises can be done in the water to reduce stress on the joint while providing resistance; strengthening of specific muscle groups to maximize joint stability should be prescribed. Contact sports are generally contraindicated. Joint replacement is used in end-stage arthropathy; loosening occurs more often, especially in younger children. Sickle Cell Disease Joint involvement occurs in infancy in sickle cell disease. Bones and joints are often the site of vaso-occlusive episodes, and chronic infarcts may result. One of the earliest manifestations of sickling in young children is dactylitis or “hand–foot syndrome.” An episode of painful swelling of the bones of the hand or foot may predict severe disease (126). Abnormalities of the vertebrae (“fish mouthing”) are characteristic of sickle cell disease. Hyperplasia of the bone marrow may cause growth disturbances and osteopenia. Osteomyelitis is also more common and may be difficult to distinguish from infarction; radionucleotide imaging and bone aspiration are often necessary to diagnose bone infection. Multiple joints can be involved in septic arthritis caused by S. aureus, E. coli, Enterobacter, and Salmonella. More often, noninflammatory joint effusions of the knee, ankle, or elbow occur during crises. Chronic synovitis in wrists, metacarpal heads, and calcanei with resultant erosive joint destruction has been reported in children with sickle cell disease. Avascular necrosis of the femur and, less often, the humeral head and TMJ can occur in sickle cell anemia (127,128). Avascular necrosis of the weight-bearing joints (hip and shoulders) causes chronic pain and may require surgical intervention. Plain x-ray films may not detect early disease, and MRI may be necessary. Early disease may improve with coring and osteotomy (129). Late disease requires joint replacement. Patients with sickle cell disease have an increased incidence of infection and failure of prosthesis. Ischemic stroke is one of the most devastating problems in children. The optimal setting for the care of patients with sickle cell disease is a comprehensive center, with a multidisciplinary team to provide ongoing support. Summary The management of children and adolescents with chronic rheumatic disease is broad and multidisciplinary (119). Pediatric physiatrists can help provide supportive treatment to children with rheumatic disease by prescribing appropriate pain medications, exercise, bracing, and equipment to maintain or restore age-appropriate function and development. Such treatment can help prevent deformity and contractures; promote normal growth; and maximize physical, psychosocial, and cognitive development in children with rheumatic disease.
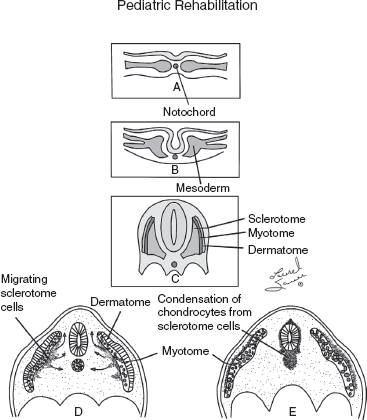
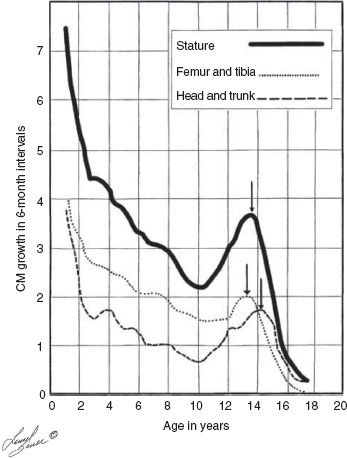
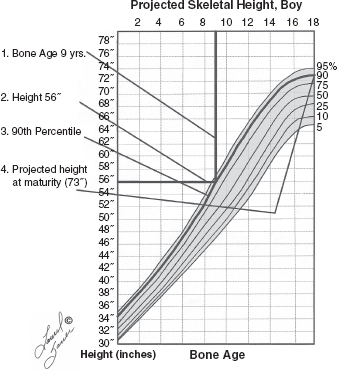
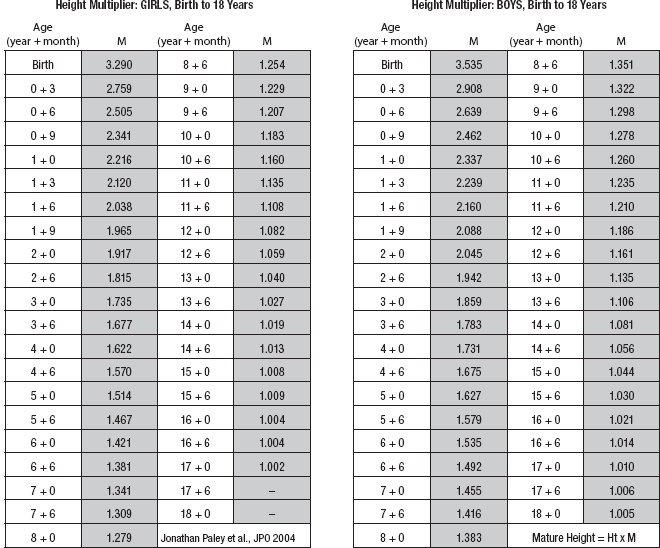
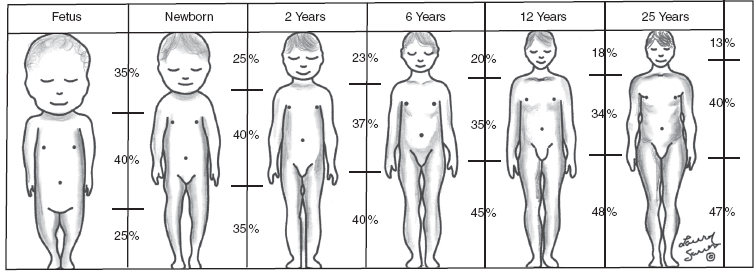
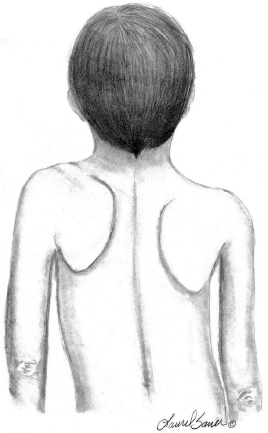
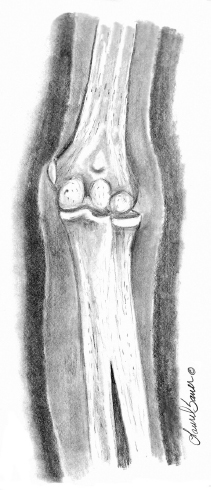
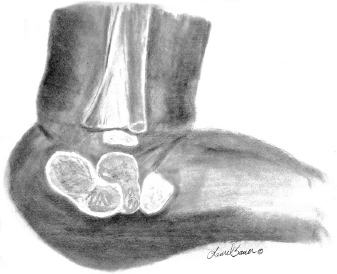
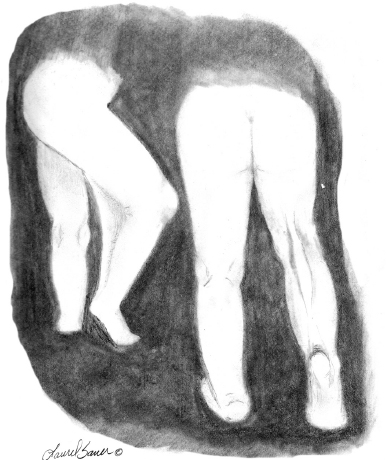
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


