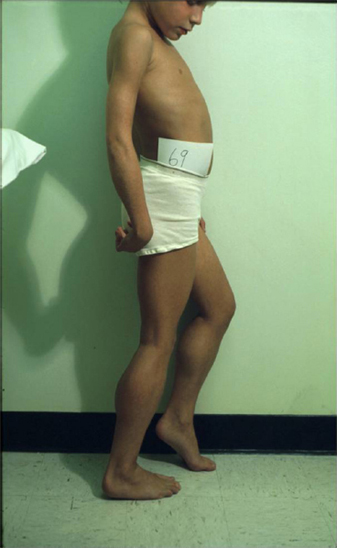
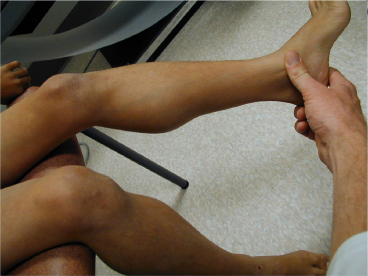
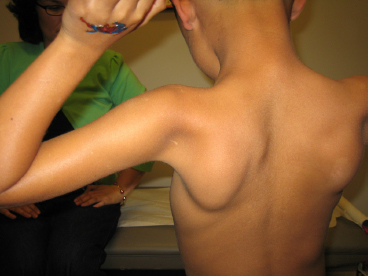
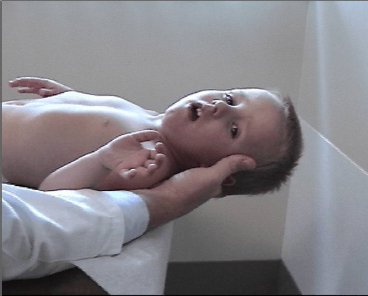
18 NEUROMUSCULAR DISEASES Craig M. McDonald INTRODUCTION Acquired or hereditary neuromuscular diseases (NMDs) are disorders caused by an abnormality of any component of the lower motor neuron—anterior horn cell, peripheral nerve, neuromuscular junction (NMJ; presynaptic or postsynaptic region), or muscle. These diseases affect children and adults with variable onset over the life span and they are often progressive with variable severity and rates of progression. While some NMDs have pathologic abnormalities isolated to one anatomic region of the lower motor neuron, with primary or secondary changes in muscle, other NMDs have been recognized as multisystem disorders. For example, myotonic muscular dystrophy may affect skeletal muscle, smooth muscle, myocardium, brain, and ocular structures; Duchenne muscular dystrophy (DMD) gives rise to abnormalities of skeletal and cardiac muscle, the cardiac conduction system, and brain; Fukuyama congenital muscular dystrophy (CMD) affects skeletal muscle and brain; mitochondrial encephalomyopathies may affect the mitochondria of multiple tissues. Appropriate rehabilitation management of NMDs requires an accurate diagnosis. All diagnostic information needs to be interpreted, not in isolation but within the context of relevant historical information, family history, physical examination findings, laboratory data, molecular diagnostic studies, electrophysiologic findings, and pathologic information, if obtained. The features of the diagnostic evaluation of NMDs pertinent for the physiatrist and neuromuscular medicine specialist have recently been reviewed (1). A skilled synthesis of all available information may provide the patient and family with (a) a precise diagnosis or as accurate a diagnosis as is medically possible; (b) prognostic information (if available for a specific entity); and (c) anticipatory guidance for the near future. Knowledge of the natural history of specific NMD conditions helps in the ongoing rehabilitative management of progressive impairments in body structure and body function, reduced activities, disabilities, and handicap. This chapter summarizes the diagnostic features, natural history and impairment profiles, and rehabilitation management of the most common childhood NMDs. SPECIFIC NEUROMUSCULAR DISEASE CONDITIONS DYSTROPHIC MYOPATHIES “Muscular dystrophies” are debilitating myopathic disorders that present with muscle wasting and diffuse muscle weakness. They are caused by genetic mutations which produce muscle fiber necrosis and regeneration, ultimately resulting in muscle fiber loss. They are characterized by elevated serum creatine kinase (CK), and muscle biopsies typically show variation in fiber size, muscle necrosis, and increased amounts of fat and connective tissue. Most types of muscular dystrophy are not purely muscle disorders, but multisystem disorders with disease manifestations in a variety of body systems, which may include the musculoskeletal, cardiovascular, pulmonary, and gastrointestinal systems, as well as endocrine system, skin, eyes, brain, and other organ systems. Muscular dystrophies are caused by mutations of the genes encoding for structural proteins which are located either in the subsarcolemmal region, within the sarcolemmal membrane, or in the extracellular region. These proteins are important for the stability of the sarcolemmal membrane and the maintenance of muscle fiber intracellular homeostasis. They are genetically, biochemically, and clinically diverse diseases. In healthy muscle, muscle fiber damage leads to a moderate activation of connective tissue cells. Satellite cells are activated and replace the damaged muscle with new muscle fibers. In muscular dystrophy, the primary defect (eg, dystrophin deficiency) leads to continuous myofiber degeneration with massive activation of connective tissue cells, resulting in fibrosis. The muscle regeneration is unable to keep pace in order to replace the damaged muscle fibers with new muscle fibers. Dystrophinopathies DUCHENNE MUSCULAR DYSTROPHY. Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder caused by a genetic mutation in the dystrophin gene at the Xp21 gene loci which codes for the protein dystrophin. The dystrophin gene which causes DMD and Becker muscular dystrophy (BMD) has a coding sequence that contains 79 exons. The primary protein product, dystrophin is localized to the intracellular side of the plasma membrane of all skeletal muscle cells, and other isoforms of dystrophin are located in certain types of neurons, and in cardiac muscle cells, the Purkinje cells of the cardiac conduction system, and smooth muscle cells of the gastrointestinal tract (2). Dystrophin deficiency at the plasma membrane of muscle fibers disrupts the membrane cytoskeleton and leads to the secondary loss of other components of the muscle cytoskeleton. Dystrophin links the intracellular actin cytoskeleton to the sarcolemmal membrane and serves as a shock absorber for the muscle cells and the primary consequence of dystrophin deficiency is membrane instability, leading to membrane injury from mechanical stresses, transient breaches of the membrane, membrane leakage, and a cascade of events subsequent to loss of homeostasis. Chronic dystrophic myopathy is characterized by aggressive fibrotic replacement of the muscle and eventual failure of regeneration with muscle fiber death and fiber loss. Generally loss of the open reading frame causes complete absence of dystrophin and a Duchenne phenotype. For cases with a deletion mutation, the “reading frame” hypothesis predicts that BMD patients with in-frame deletions produce a semifunctional, internally deleted dystrophin protein, whereas DMD patients with frameshift or “out-of-frame deletions produce a severely truncated protein that would be unstable and not persist (3). Characteristics of DMD and BMD are shown in Table 18.1. Consensus care considerations for DMD using expert opinions and RAND methodology have been published (4,5). Diagnostic Evaluation. Data from the Centers for Disease Control and Prevention (CDC) Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) among 156 boys without a known family history of DMD prior to birth, first signs, or symptoms were noted at a mean age of 2.5 years. Concerns resulted in primary care provider evaluation of the child at a mean age of 3.6 years. The mean age at the time of initial CK was 4.7 years. The mean age at definitive diagnosis of DMD was 4.9 years. Thus, there is a delay of about 2.5 years between the onset of DMD symptoms and the time of definitive diagnosis, unchanged over the previous two decades (6). This delay results in lost opportunities for timely genetic counseling and initiation of corticosteroid treatment. It is recommended that clinicians check the CK level early in the evaluation of boys with unexplained developmental delay as serum CK is a useful screening test for DMD and a normal CK value rules out DMD. Approximately 60% of mutations are large-scale deletions, 5% are duplications, and the remainder, detectable from genomic DNA, are point mutations or small deletions/insertions. The remaining ~5% of mutations are due to intronic mutations that are undetectable by standard genomic analysis but result in altered splicing only detectable by messenger ribonucleic acid (mRNA) analysis from muscle tissue. Thus, lack of a detectable DMD gene mutation using standard methodology does not exclude a DMD diagnosis. Muscle biopsy and dystrophin expression analysis remains the gold standard of diagnosis, remaining particularly useful in cases where no mutation was detected by standard clinical molecular diagnostic testing. Full gene sequencing is now the standard of care in all patients at risk of a dystrophinopathy, and in the United States funding is available for detailed molecular diagnostics if insurance coverage is insufficient. This testing is both for diagnostic purposes and to identify candidates for future molecular-based therapies such as exon skipping with oligonucleotides or morpholinos, nonsense-mediated suppression therapy for the 12% to 15% of patients with DMD and BMD with premature stop codon mutations, and other novel therapies that will require knowledge of specific gene sequence alterations. In patients suspected of dystrophinopathy with no family history and molecular genetics which do not clearly differentiate a DMD and BMD phenotype, a muscle biopsy with immunostaining and quantitative dystrophin analysis with Western blot or novel quantitative dystrophin assays using immunostaining or mass spectrometry is critical to allow patients to be eligible for future clinical trials of novel therapeutics with rigid inclusionary criteria. Immunohistochemical, immunofluorescent, or Western blot analysis can show the relative amount of dystrophin in skeletal muscle specimen, and Western blot can reveal its size, helping to distinguish between DMD and milder muscular dystrophy phenotype such as BMD. An amount of dystrophin of less than 3% to 5% of normal has been described as consistent with DMD, and greater than 20% as consistent with BMD, but standardization of dystrophin quantification is challenging, and in current clinical diagnostic practice, dystrophin expression is frequently descriptive or semiquantitative. Intermediate DMD may be characterized by a dystrophin quantity greater than 5%, ambulation past 12 years (if steroid naïve), and 14 years (if treated with steroids). Epidemiology and Survival. The incidence of DMD, based on a number of population studies as well as neonatal screening has been estimated to be around 1:3,500 to 5,500 male births (7,8). As many as one-third of isolated cases may be due to new mutations, which is considerably higher than observed in other X-linked conditions. This high mutation rate may relate to the large size of the gene. There has been a changing natural history in DMD over the past four decades affecting survival from the 1960s when there was largely no supportive treatment to the 1970s and 1980s when survival was improved by spinal surgery and earlier provision of noninvasive ventilation (9,10). Provision of noninvasive ventilation has been recognized as the main intervention affecting survival (9,11) with ventilated mean survival increasing from 17.7 years to 27.9 years in one study (9) and 19.0 years to 27.0 years in another study (11). In addition, survival in DMD has been enhanced by glucocorticoids (12) and angiotensin-converting enzyme (ACE) inhibitors (13). TABLE 18.1 CHARACTERISTICS OF DYSTROPHINOPATHIES (DMD AND BMD) DMD BMD U.S. prevalence (est.) 15,000 2,200 Incidence rate 1/3,500 to 5,500 male births Unknown Inheritance X-linked X-linked Gene location Xp21 (reading frame shifted) Xp21 (reading frame maintained) Protein Dystrophin Dystrophin Onset 2–6 years 4–12 years (severe BMD) Late teenage to adulthood (mild BMD) Severity and course Relentlessly progressive • Reduced motor function by 2–3 years • Steady decline in strength • Life span < 35 Slowly progressive • Severity and onset correlate with muscle dystrophin levels Ambulation status • Loss of ambulation: 7–13 years (no corticosteroids) • Loss of ambulation 9–16 years (corticosteroids) • Loss of ambulation: >16 years Weakness Proximal > distal • Symmetric • Legs and arms (lower extremity weakness predates upper extremity weakness by approximately 2 years) Proximal > distal • Symmetric • Legs and arms Cardiac Dilated cardiomyopathy first to second decade Onset of signs second decade Cardiomyopathy (may occur before weakness); third to fourth decade frequent Respiratory • Profoundly reduced vital capacity in second decade • Ventilatory dependency in second decade • Respiratory involvement in subset of patients • Ventilatory dependency in severe patients Muscle size • Calf hypertrophy • Calf hypertrophy Musculoskeletal • Contractures—ankles, hip, and knees • Scoliosis: onset after loss of ambulation • Contractures: ankles and others in adulthood Central nervous system • Reduced cognitive ability in some • Reduced verbal ability • Minority of patients have reduced cognitive ability Muscle pathology • Endomysial fibrosis and fatty infiltration • Variable fiber size and • Myopathic grouping • Fiber degeneration/regeneration • Dystrophin: 3% or less • Sarcoglycans: secondary reduction • Variable fiber size • Endomysial connective tissue and fatty infiltration • Fiber degeneration • Fiber regeneration • Dystrophin: reduced (usually 20%–60% of normal) Blood chemistry and hematology • Newborn CK usually > 2,000 CK: Very high (10,000–50,000) by 2 years of age • High AST and ALT (normal GGT) • High aldolase • CK: 5,000 to 20,000 • Lower levels of CK with increasing age Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transferase. Onset and Early Signs. While the history of hypotonia and delayed motor milestones are often reported in retrospect, the parents are often unaware of any abnormality until the child starts walking. There has been variability reported in the age of onset (14,15). In 74% to 80% of instances, the onset has been noted before the age of 4 years (14–16). The vast majority of cases are identified by 5 to 6 years of age. In addition to gross motor delays, cognitive and language delays have also been documented prior to the age of 3 in DMD (17,18). The most frequent presenting symptoms have been abnormal gait, frequent falls, and difficulty climbing steps. Parents frequently note the toe walking, which is a compensatory adaptation to knee extensor weakness and a lordotic posture to the lumbar spine, which is a compensatory change due to hip extensor weakness (Figure 18.1). Occasionally, DMD is identified presymptomatically in situations where a CK value is obtained with a markedly elevated value, malignant hyperthermia occurs during general anesthesia for an unrelated surgical indication, or a diagnosis is pursued in a male with an affected older sibling. Difficulty negotiating steps is an early feature as is a tendency to fall due to the child tripping or stumbling on a plantar-flexed ankle or the knee buckling or giving way due to knee extensor weakness. There is progressive difficulty getting up from the floor with the presence of a Gower’s sign (Figure 18.2). Pain in the muscles, especially the calves, is a common symptom. Enlargement of muscles, particularly the calves (Figure 18.3), is commonly noted. The deltoid may also be hypertrophied. With the patient’s arms abducted to 90 degrees and externally rotated, the hypertrophy of the posterior deltoid and infraspinatus frequently leaves a depression between these two muscles referred to as the “posterior axillary depression sign” in DMD (Figure 18.4). The tongue is also frequently enlarged. There is also commonly an associated wide arch to the mandible and maxilla with separation of the teeth, presumably secondary to the macroglossia. FIGURE 18.1 “Myopathic” stance in an 8-year-old male with Duchenne muscular dystrophy. Notice the lumbar lordosis to compensate for hip extensor weakness and primarily forefoot contact to compensate for knee extensor weakness. Pattern and Progression of Weakness. The earliest weakness is seen in the neck flexors during preschool years (Figure 18.5). Weakness is generalized but predominantly proximal early in the disease course. Pelvic girdle weakness predates shoulder girdle weakness by several years. Ankle dorsiflexors are weaker than ankle plantar flexors, ankle everters are weaker than ankle inverters, knee extensors are weaker than knee flexors, hip extensors are weaker than hip flexors, and hip abductors are weaker than hip adductors (15). The weakness progresses steadily, but the rate may be variable during the disease course. Quantitative strength testing shows greater than 40% to 50% loss of strength by 6 years of age (15). With manual muscle testing, DMD subjects exhibit loss of strength in a fairly linear fashion from ages 5 to 13 and measurements obtained several years apart will show fairly steady disease progression. A variable course may be noted when analyzing individuals over a shorter time course (15). Quantitative strength measures have been shown to be more sensitive for demonstrating strength loss than manual muscle testing when strength is graded 4 to 5 (15). FIGURE 18.3 Calf pseudohypertrophy in a male with Duchenne muscular dystrophy. Disease Progression and Loss of Milestones. What follows is a brief overview of the current natural history of DMD across the spectrum of disease. Furthermore, while specific functional changes are observed at different ages, it should be emphasized that the disease is due to generalized skeletal muscle involvement, and cardiomyopathy and pathologic processes involved in DMD are ongoing over the course of a patient’s lifetime — while loss of ambulatory capacity and gross motor functions may be a primary focus in ambulatory boys, neuromuscular deterioration may already be measurable in the upper limb and other muscle groups. Note, the ages are approximations, and the intent is not to create artificial stages of the disease. FIGURE 18.4 Posterior axillary depression sign in Duchenne muscular dystrophy. Note the prominent deltoid superolaterally and infraspinatus inferomedially. FIGURE 18.5 Weakness of neck flexors in an 8-year-old with Duchenne muscular dystrophy makes it difficult for him to bring his chin to the chest when supine and to hold his head up when placed at the end of the examination table. • Neonates/Infancy: While DMD is rarely diagnosed in infancy, the disease is manifested at birth. Even though some of the infants detected due to family history are sometimes referred to as being asymptomatic, most will still show delayed development if evaluated with tools such as the Griffiths Mental Development Scales, an outcome measure than can be used in the very young (6–47 months) and the Bayley Scales of Infant and Toddler Development, Third Edition (Bayley-III). One study of children with DMD with mutations upstream or in exon 44 had higher Developmental Quotient (DQ) than those with mutations downstream exon 44 which are associated with involvement of dystrophin isoforms expressed at high levels in the brain. The difference was significant for total and individual subscale DQ with the exception of the locomotor subscale. Items, such as ability to run fast, or getting up from the floor consistently failed in all children, irrespective of the age or of the site of mutation. • Young children, early ambulant (aged 1 to 42 months): The development of gross motor milestones is typically slower than in boys without Duchenne, and some children may show signs of delayed language and cognitive impairment. Toddlers and young children may also be scored with developmental outcome measures such as the Bayley-III and Griffith’s Developmental Scales. Gross motor scores were lower in young children with DMD at baseline compared with published controls and revealed a further declining trend at 6 months. Repeated measures analysis over 12 months revealed that gross motor scores declined further at 12 months. Cognitive and language scores were lower at baseline compared with typically developing children and did not change significantly at 6 or 12 months. Fine motor skills, also low at baseline, improved over 1 year. • Young ambulatory (from 4 to approximately 7 years): A period where there may be slower gains in ambulatory function as compared to typically developing children (on 6-minute walk test (19–22) and 10-meter walk/run test) and either gains or losses in milestones as noted by end points such as the North Star Ambulatory Assessment (NSAA) (23). However, it is important to note that physiologic deterioration is ongoing and boys are increasingly falling behind normative performance levels of their normally functioning peer group. • Late ambulatory (from approximately seven to thirteen years of age): Generally defined as when individuals begin to suffer a decline in their gross motor functions as well as some pulmonary function parameters, particularly maximal expiratory pressure (MEP) and maximal inspiratory pressure (MIP). During this stage of disease, there is marked progressive loss of muscle fiber in the proximal muscles, growing weakness, and the gradual loss of gross motor skills and ambulatory functions (including standing ability, stair climbing, and ultimately, the ability to walk). The six-minute walk distance (6MWD) typically declines after age 7, and a 30-meter decline in 6MWD in comparison to stability is predictive of a greater tendency to lose ambulation over the following 2 years. The average age of full-time wheelchair use in a DMD population not treated with corticosteroids has been age 10 with a range of 7 to 13 (15). Timed motor performance is useful for the prediction of time when ambulation will be lost without provision of long-leg braces. The 10-meter walk/run test has been shown to be predictive of subsequent loss of ambulation. Two large natural history studies have showed that a large proportion of DMD subjects who took 12 seconds or longer to ambulate 10 meters or approximately 30 feet lost ambulation within 1 year (15,22). Ambulation past the age of 14 in a non-cortico-steroid-treated patient should raise the suspicion of a milder form of muscular dystrophy such as BMD or limb girdle muscular dystrophy (LGMD). Ambulation beyond 16 years has been previously used as an exclusionary criterion for DMD in studies of BMD. Immobilization for any reason can lead to a marked and often precipitous decline in muscle power, rapid development of contractures, and loss of ambulatory ability. A fall with resultant fracture leading to immobilization and loss of ambulatory ability is not an uncommon occurrence. Ankle equinus contractures are the most common skeletal deformity. There is risk of osteopenia and fractures. There is also a comparative loss in height and increased weight gain in comparison to their normally functioning peer group. • Early nonambulatory (beginning at the age when a boy starts using a wheelchair full time): After boys can no longer walk, there is continued muscular deterioration throughout the upper and lower limbs, and skeletal deformities such as lower and upper limb contractures and spine deformity may become problematic. Powered mobility is required after loss of ambulation. Postural maintenance and sitting balance are initially intact and progressively lost. There is increasing loss of upper limb function (with decreasing ability to reach overhead, dress, self-feed, and perform other self-care). There is continued decline in pulmonary function with the ultimate need for mechanical cough assistance and progressive risk of nocturnal hypoventilation requiring noninvasive ventilation. Cardiomyopathy is evident by cardiac MRI in virtually all patients and in some patients by cardiac echo. After transition to a wheelchair, patients tend to put on more weight compared to their normally functioning peer group. • Late nonambulatory: Postural support of the trunk and head support from a seating system are required as well as power recline. Upper extremity function is severely limited to distal fine motor function and tabletop activities. Maintaining computer access is a critical quality of life concern. Virtually all patients benefit from mechanical cough assistance and there is a high risk of nocturnal and daytime hypoventilation requiring noninvasive ventilation. Optimal nutritional management may require gastrostomy tube placement and enteral formula supplementation. There is risk for dysphagia and aspiration. Adequate phonation may become an issue late in the disease course. There may be a larger number of older DMD patients with unmet medical needs. As patients age, respiratory impairment and heart disease (heart failure and conduction abnormalities) are causes of morbidity and, eventually, mortality.
CK usually < 5,000 by second decade

Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


