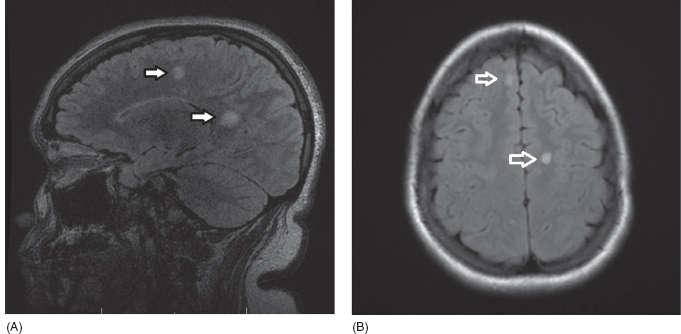
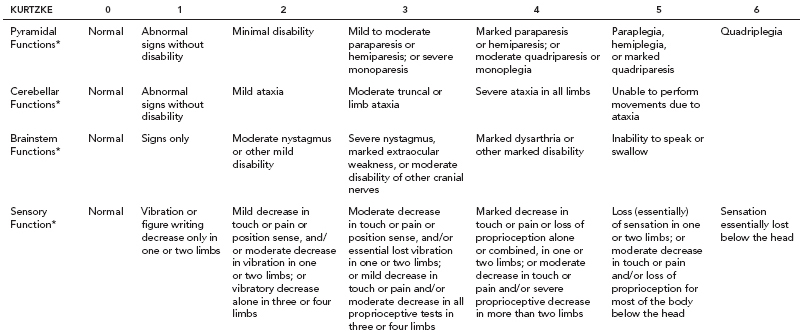
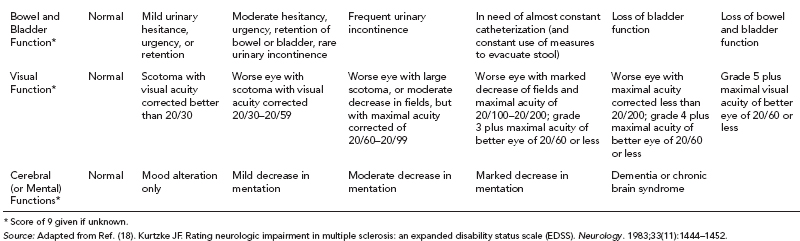
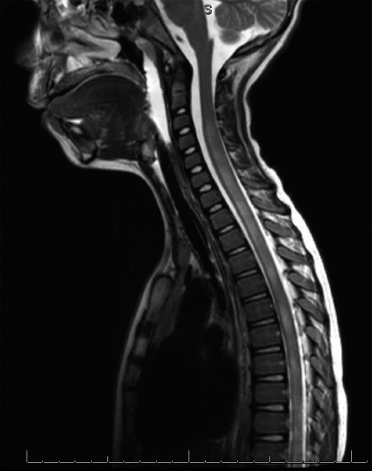
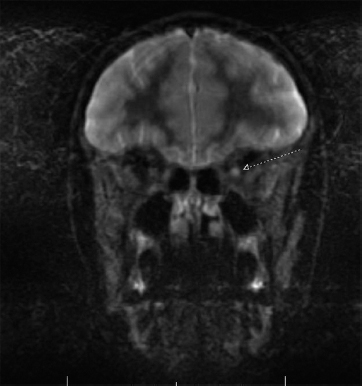
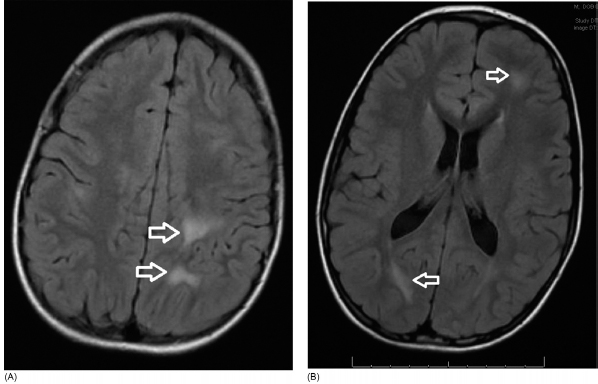
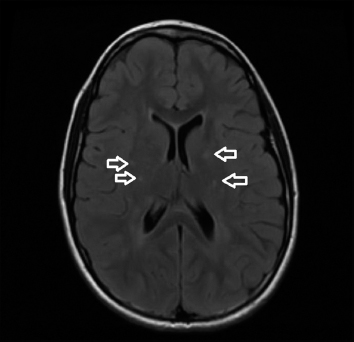
19 NEURODEGENERATIVE AND DEMYELINATING DISEASES AND OTHER CNS DISORDERS Amy Houtrow This chapter covers numerous conditions that pediatric rehabilitation medicine physicians may encounter during their training and career. Because data contained in other chapters of this book are particularly relevant to the conditions presented here, the reader will be guided back to other chapters from time to time. This chapter will address pediatric demyelinating diseases of the central nervous system (CNS) and acquired brain injury from encephalitis (including anti-N-methyl-D-aspartate receptor [NMDAR] encephalitis), pediatric brain tumors, and seizure focus resection. PEDIATRIC DEMYELINATING DISEASES In this section of this chapter, the following pediatric demyelinating diseases of the CNS will be described: multiple sclerosis (MS), transverse myelitis, neuromyelitis optica (NMO), and acute disseminated encephalomyelitis (ADEM). Together, these conditions have incidence in children of 1.66 per 100,000 person-years (1). They are more common among Whites and Blacks and less common among Hispanics and Asians (2). Acute inflammatory demyelinating polyradiculoneuropathy (Guillain–Barré syndrome), a peripheral process, is described in Chapter 18. Generally, demyelinating diseases are described as monophasic or polyphasic and monofocal or polyfocal. Monophasic disease is also often referred to as a clinically isolated syndrome (3). Some children with monophasic disease go on to develop chronic demyelinating conditions such as MS or NMO. And it is important to note that there is considerable clinical overlap between these conditions (4). PEDIATRIC MULTIPLE SCLEROSIS Multiple sclerosis (MS) is an autoimmune chronic inflammatory disease characterized by demyelination and axonal degeneration. Approximately 5% of the total population of individuals with MS are children (5). In 2007, the International Pediatric Multiple Sclerosis Study Group proposed provisional definitions for CNS demyelinating disorders for children that were updated in 2012 and published in 2013 (6). The diagnosis requires the presence of CNS inflammatory disease that is disseminated in time and space. Pediatric MS can be diagnosed when any one of the following exists: • Two or more nonencephalopathic clinically identified CNS events with presumed inflammatory cause, which are separated by more than 30 days and involve more than one area of the CNS • One nonencephalopathic episode with associated MRI findings consistent with the revised 2010 McDonald criteria (7) (see Table 19.1), and follow-up MRI demonstrates one or more lesions consistent with the disseminated in time criteria • One encephalopathic (ADEM) attack followed by a nonencephalopathic attack three or more months after the onset of symptoms with MRI lesions that meet the disseminated in space criteria • For children 12 years old or older, a single nonencephalopathic event with MRI findings consistent with the criteria for dissemination in time and space EPIDEMIOLOGY OF PEDIATRIC MS While 1.7% to 5.6% of the total MS population is under the age of 18, diagnosis under the age of 10 is very rare. The incidence of pediatric MS worldwide is not known, but in California, the reported incidence is 0.51/100,000 person-years (1). There is greater racial/ethnic diversity among children with MS compared to adults, among whom most are non-Hispanic Whites (5). The remote history of Epstein–Barr virus is associated with pediatric MS and many studies have supported the geneviral environment hypothesis (7). The exact mechanisms that lead to MS are not understood and ongoing research is being conducted to identify triggers and the pathophysiology. TABLE 19.1 2010 MCDONALD MRI CRITERIA FOR THE DIAGNOSIS OF MULTIPLE SCLEROSIS DEMONSTRATION OF DISSEMINATION IN SPACE DEMONSTRATION OF DISSEMINATION IN TIME At least two of four areas of the CNS with one or more T2 lesions • Periventricular • Juxtacortical • Infratentorial • Spinal cord (excluded if patient has a brainstem or spinal cord syndrome) 1. One or more new T2 and/or gadolinium-enhanced lesion(s) on follow-up MRI in comparison to baseline scan 2. Simultaneously present gadolinium-enhancing asymptomatic and nonenhancing lesions at any time as long as the asymptomatic gadolinium-enhancing lesion is not due to non-MS pathology Source: Adapted from Ref. (7). Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302. CLINICAL COURSE OF PEDIATRIC MS AND DIAGNOSIS Almost uniformly, children with MS have relapsing–remitting type (3). The rate of relapses in children are higher than adults with MS, but recovery seems to occur more quickly (3). Frequently, children are polysymptomatic at presentation. Common presenting symptoms include sensory deficits, optic neuritis, gait disturbances, and brainstem symptoms (3). There is clinical overlap with ADEM raising concern that children who present with ADEM may eventually meet criteria for MS. In addition, children with MS have cognitive deficits more commonly than adults. Thirty-five percent of consecutively enrolled subjects from the Pediatric Multiple Sclerosis Centers for Excellence met diagnostic criteria for cognitive impairment (8). When MS is suspected, a complete neurologic exam is necessary as is MRI with gadolinium of the brain and spinal cord. MRI findings consistent with MS include ovoid T2 lesions, and fluid-attenuated inversion recovery (FLAIR) hyperintensities in the periventricular white matter are typical. Cortical lesions are less common in children with MS compared to adults (9). See Figure 19.1 for an MRI of a teenaged girl with MS. FIGURE 19.1 The sagittal (A) and axial (B) views of the MRI of a teenaged girl with MS demonstrate T2-FLAIR hyperintensities separated in space. The arrows point to the lesions. Source: Images courtesy of Guilio Zuccoli, MD, Associate Professor of Radiology, University of Pittsburgh. Children with MS experience two to three times more frequent relapses than adults with an annualized relapse rate of 1.12 to 2.76 (10). Their MRI lesion burden is higher than adults, as well (11). Additionally, they have higher rates of cognitive impairments and longitudinally demonstrate worsening of cognitive functioning (12). Conversely, children with MS accrue locomotor disability more slowly than adults (13). At the time of disease onset, a progressive initial course is predictive of a statistically shorter time to irreversible disability (13). Two years after onset, the number of relapses was predictive of increased rate of disability as was having a progressive initial course (13). TREATMENT FOR PEDIATRIC MS The International Pediatric Multiple Sclerosis Study Group recommends beta-interferon or glatiramer acetate as first-line therapy for pediatric MS (14). In the presence of inadequate treatment response, defined as a 6-month minimum of full dose fully adherent therapy with either persistent or increased relapse rate, or two or more confirmed relapses in 12 months, the study group recommends switching between first-line agents or switching to or adding a second-line agent (14). High-dose steroids are often used for short periods and natalizumab and cyclophosphamide are also second-line options (15,16). Some providers use mitoxantrone, fingolimod, or rituximab as second-line options although the evidence is more limited regarding their use (14,17). REHABILITATION FOR PEDIATRIC MS Since the 1980s, the Expanded Disability Status Scale (EDSS) and the Functional Systems Score (FSS) have been used to track MS-related disability (18). The systems in the FSS are: pyramidal functions, cerebellar functions, brainstem functions, sensory function, bowel and bladder function, visual function, and cerebral (mental) functions (18). See Table 19.2 for the FSS. The scores on the EDSS range from 0 (normal neurologic exam) to 10 (death due to MS) and focus on mobility skills and FSS dysfunction. See Table 19.3 for the complete EDSS. The EDSS does not map well to functional assessment tools typically used in pediatric rehabilitation such as the Functional Independence Measure for Children (WeeFIM), Pediatric Evaluation of Disability Inventory (PEDI), and Functional Rehabilitation Evaluation of Sensori-Neurologic Outcomes (FRESNO) (19–21). Nonetheless, the EDSS is used in most research related to the clinical management of MS and is important for the rehabilitation physician to understand. Developmentally speaking, EDSS is not appropriate for children who would not be expected to be ambulatory or independent with their self-care. This is rarely a problem because MS is extremely uncommon among very young children. The rehabilitation interventions depend on the functional impairments, activity limitations, and participation restrictions experienced by the child with MS. Common impairments include: weakness, spasticity, neurogenic bladder, neurogenic bowel, pain, fatigue, and depression. Although physical and occupational therapy are the primary way weakness is addressed, potassium channel blockade has been shown to improve ambulation due to the improved conduction in demyelinated fibers (22). Because of the narrow therapeutic window, it is not recommended for use in children. When considering an antispasticity agent such as baclofen, it is important to evaluate if the spasticity is benefiting function such as for ambulation when spasticity in the lower extremities is compensating for weakness (23). It is also important to consider the side effect of fatigue because fatigue is a common symptom of MS and may be made worse with the initiation of an antispasticity agent. For intractable spasticity, intrathecal baclofen pump placement may be a consideration. For more localized spasticity, botulinum toxin injection should be considered (24). Botulinum is also used for neurogenic detrusor overactivity (25). But the first-line management remains anticholinergic agents such as oxybutynin (23). Symptomatic neurogenic bladder impacts about 80% of all patients with MS with detrusor hyperactivity and detrusor-sphincter dyssynergia as the most common diagnoses (23). There is little consensus about when to start intermittent straight catheterizations (26), but issues regarding continence should be discussed both from the medical and social perspectives. For the adolescent with MS, discussions about sexual functioning should occur. Management of neurogenic bowel and constipation follows the recommendations for management of upper motor neuron bowel (see Chapter 16 for details). Neuropathic pain and musculoskeletal pain are common. Antiepileptic medications play an important role in the genic bladder impacts about 80% of all patients with MS with detrusor hyperactivity and detrusor-sphincter dyssynergia as the most common diagnoses (23). There is little consensus about when to start intermittent straight catheterizations (26), but issues regarding continence should be discussed both from the medical and social perspectives. For the adolescent with MS, discussions about sexual functioning should occur. Management of neurogenic bowel and constipation follows the recommendations for management of upper motor neuron bowel (see Chapter 16 for details). Neuropathic pain and musculoskeletal pain are common. Antiepileptic medications play an important role in the modulation of neuropathic pain and traditional analgesics are helpful in musculoskeletal pain (7). In adults, the use of cannabis extract has been used with success in the management of painful spasticity refractory to other interventions (27). When a child presents with debilitating fatigue, a close examination of the medications should be conducted to determine whether they are contributing to the fatigue. A sleep hygiene regime may be initiated and medications modafinil and amantadine may be considered, but their efficacy is limited (23). Mood disorders are frequent among individuals with MS and likely under-recognized in children. Psychological interventions, including cognitive behavioral therapy have been shown to be helpful (23). Medications to address mood may be considered. TABLE 19.2 KURTZKE FUNCTIONAL SYSTEMS SCORES TABLE 19.3 THE EXPANDED DISABILITY STATUS SCALE KURTZKE EXPANDED DISABILITY STATUS SCALE 0 Normal neurological exam: FSS = 0 in all systems (score of 1 on cerebral function is allowable) 1 No disability: minimal signs in functional system 1.5 No disability: minimal signs in more than one functional system 2 Minimal disability: FSS = 2 in one functional system, other 0 or 1 2.5 Minimal disability: FSS = 2 in two functional systems, others 0 or 1 3 Moderate disability: FSS = 3 in one functional system, others 0 or 1, or mild disability in three to four functional systems 3.5 Fully ambulatory but FSS = 3 in one functional system and FSS = 2 in two other functional areas; FSS = 3 in two functional areas, other 0 or 1; or FSS = 2 in five functional systems 4 Fully ambulatory without assistive device, able to walk 500 m without rest, self-sufficient despite relatively severe disability: FSS = 4 in one functional system, others 0 or 1; or combination of lesser grades exceeding previous levels 4.5 Fully ambulatory without assistive device, able to walk 300 m without rest, able to work, may otherwise have some limitations of full activity or need minimal assistance: usually FSS = 4 in one functional system or combination of lesser grades exceeding previous levels 5 Ambulatory without assistive device or rest for 200 m, full day activities impaired: FSS = 5, others 0 or 1, or combination of lesser grades exceeding level 4 5.5 Ambulatory without assistive device or rest for 100 m, full day activities impaired: FSS = 5 for one functional domain or combination of lesser grades exceeding level 4 6 Intermittent or constant assistive device required to walk 100 m with or without rest: usually FSS = 3+ in more than two functional systems 6.5 Constant bilateral assistive device(s) required to walk 20 m: usually FSS = 3+ in more than two functional systems 7 Unable to walk 5 m with assistive device(s), transfers independently, can propel manual wheelchair for full day: usually combinations of functional system impairments with more than one FSS = 4+ 7.5 Unable to take more than a few steps, assistance for transfers, cannot self-propel manual wheelchair: usually combinations of functional system impairments with more than one FSS = 4+ 8 Essentially restricted to bed or wheelchair but retains many self-care functions, generally has functional use of arms: usually combinations of functional system impairments with FSS = 4+ in several systems 8.5 Essentially restricted to bed, some effective use of arms, retains some self-care skills: usually combinations of functional system impairments with FSS = 4+ in several systems 9 Dependent but can communicate and eat: usually combinations of functional system impairments with FSS = 4+ in most systems 9.5 Totally dependent: almost all FSS = 4+ 10 Death due to MS Abbreviation: FSS, Functional systems score. Source: Adapted from Ref. (18). Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(11):1444–1452. Pediatric MS is a rare and complicated condition that can impact functioning in multiple different domains. The pediatric rehabilitation physician has many resources available to help address the disability associated with MS. These include medications to address the common symptoms of MS and the coordinated goal-directed rehabilitation team. TRANSVERSE MYELITIS Transverse myelitis, also called acute transverse myelitis, is an acute monophasic or clinically isolated syndrome of the spinal cord (4). Transverse myelitis may also be associated with polyphasic condition such as neuromyelitis optica, MS, or systemic lupus erythematosus (SLE) (28). The diagnosis relies on the exclusion of other causes of myelitis and spinal cord compression. Once other etiologies are excluded, the diagnosis depends on meeting the inclusion criteria listed in Table 19.4 (29). When working up myelopathy, there are three priorities (30): 1. Ruling out compressive etiology 2. Determination of evidence of cord inflammation, and 3. Determination of the extent of the demyelination (beyond the cord or not). Epidemiology of Transverse Myelitis Transverse myelitis is a rare condition but one that often requires comprehensive inpatient rehabilitation. Approximately 20% of the 1,400 new cases in the United States each year are children (31). Children with transverse myelitis tend to be older at the age of diagnosis compared to children with ADEM (1). There appears to be a bimodal distribution with peaks in early childhood and a board peak between school-age and older children/adolescents (32). A considerable percentage of children with transverse myelitis go on to be diagnosed with a polyphasic demyelinating disorder such as MS (33). TABLE 19.4 THE DIAGNOSTIC CRITERIA FOR TRANSVERSE MYELITIS Development of dysfunction attributable to the spinal cord Bilateral signs and symptoms (not necessarily symmetric) Inflammation of the spinal cord evidenced by: • CSF pleocytosis, or • Elevated IgG index, or • Gadolinium enhancement If none are present at symptom onset, repeat MRI and lumbar puncture are warranted. Progression to nadir between 4 hours and 21 days Abbreviation: CSF, cerebrospinal fluid. Source: Adapted from Ref. (29). Transverse Myelitis Consortium Working Group. Proposed diagnostic criteria and nosology of acute transverse myelitis. Neurology. 2002;59(4):499–505. Clinical Course and Diagnosis of Transverse Myelitis If there is clinical concern for transverse myelitis, a thorough history should be gathered to document time course and extent of the symptoms, as well as any recent illness or prodromal symptoms (29). A recent mild illness is reported in a vast majority of cases; less frequently the patient may report a vaccine or allergy shot (28). The location of the lesion is most commonly the thoracic spine, but any and all segments may be involved (3,32). Therefore, a majority of patients present with complaints of lower extremity weakness. Rarely conus medullaris syndrome presentation may occur (32). In cases of posterior column involvement, fine motor discoordination and ataxic gait may mimic cerebellar disease (28). Children with posterior cord syndrome should be worked up for B12 and copper deficiency (33). In addition to weakness, most children present with constipation and bladder dysfunction consistent with neurogenic bowel and bladder, respectively. Children often also present with sensory disturbances such as allodynia and paresthesias (28,32). Horner syndrome, in lesions above T2, has also been reported (28). A full neurologic exam is warranted with delineation of motor and sensory levels. The child’s neurologic level of the injury should be determined to the best of the examiner’s ability recognizing the challenges of conducting a comprehensive spinal cord examination on a very young child. The neurologic level is the most caudal segment with normal sensation and antigravity motor function bilaterally (34). The American Spinal Injury Association impairment scale classifies the completeness of the injury based on sacral sparing, which is the presence of motor or sensory function in the most caudal sacral segments (34). The grading of the degree of impairment is important not only to anticipate function, but also because partial transverse myelitis is associated with a higher likelihood of future diagnosis of a polyphasic demyelinating disorder (31,33). In the very early phases of the disease, the child may be in spinal shock and have absent reflexes. Guillain–Barré Syndrome can present similarly with diminished reflexes, dysautonomia, and bowel and bladder dysfunction, but usually is quickly distinguished from transverse myelitis (28). See Table 19.5 for the differential diagnosis of myelitis (28,30,33,34). TABLE 19.5 THE DIFFERENTIAL DIAGNOSIS OF TRANSVERSE MYELITIS DIAGNOSTIC GROUP SPECIFIC DIAGNOSES (NONEXHAUSTIVE LISTS) Compressive myelopathy from extramedullary mass effect (excluding neoplasms) • Vertebral body compression • Disk herniation • Abscess • Epidural hematoma Neoplasm • Extramedullary: • Ewing sarcoma • Lymphoma • Neuroblastoma • Granulocytic sarcoma • Metastatic disease • Intramedullary: • Astrocytoma • Ependymoma • Glioma • Hemangioblastoma • Metastatic disease Paraneoplastic (exceptionally rare in children) • Lung cancer • Breast cancer • Ovarian cancer Toxin • Radiation associated myelopathy (usually presents within 15 years after radiation) Infectious • Viral: • DNA viruses (herpes) • RNA viruses: • Flaviviruses (West Nile) • Orthomyxoviruses (influenza A) • Paramyxoviruses (measles) • Picornaviruses (coxsackie) • Bacterial • Fungal • Parasitic Ischemic/hemorrhagic • Anterior spinal artery syndrome—presents with motor and spinothalamic-related sensory deficits with sparing of proprioception and vibratory senses (34) • Surfer’s myelopathy • Fibrocartilaginous embolism • Arteriovenous malformation Metabolic • Copper deficiency • B12 deficiency Autoimmune • Systemic lupus erythematosus • Sjogren syndrome • Mixed connective tissue disorder • Scleroderma • Neurosarcoidosis • Behcet’s disease Source: Image courtesy of Guilio Zuccoli, MD, Associate Professor of Radiology, University of Pittsburgh. A gadolinium-enhanced MRI of the spine should be conducted as soon as possible. Depending on the timing, a brain MRI can be conducted simultaneously or at a later time. If a compressive myelopathy is identified, then urgent surgical referral is warranted and steroids may be considered (29). The MRI of transverse myelitis usually reveals T1-isointense and T2-hyperintense signals over multiple contiguous segments (3,33). The entire cord may be involved and there can be substantial swelling with effacement (3). See Figure 19.2 for the MRI of a child with tetraplegia from transverse myelitis. Children with transverse myelitis tend to have more severe presentations than adults (30). Complete recovery can occur, but occurs in a minority of children (30). A number of factors have been identified as poor prognostic indicators, including: rapidity to nadir (in some studies), need for ventilator support, higher lesion level, larger number of segments involved, and spinal shock (28,30,32). Symptom nadir tends to occur within a week of presentation. Around that time, children with transverse myelitis may experience autonomic instability with fluctuations in heart rate, temperature, and respirations (28). Recovery is most rapid during the first 6 months after onset, but improvements have been documented years after the disease (30). As mentioned previously, some children, especially those with partial lesions will go on to develop a polyphasic demyelinating condition despite good recovery from the first lesion. Treatment for Transverse Myelitis High-dose steroids given for 5 to 7 days are the mainstay of treatment for transverse myelitis (3), although there is insufficient evidence to determine the efficacy of steroids (31). In fulminant disease, plasmapheresis and intravenous immunoglobulin (IVIg) may be considered (3,31). Many children present with pain and sensory changes, which may persist long-term. Medications to manage spasticity and pain are commonly needed. Children with cervical lesions may need ventilatory support. Rehabilitation for Transverse Myelitis The rehabilitation management of the child with transverse myelitis is similar to the management of children with spinal cord injuries of other etiologies (see Chapter 16). Consultations with therapies and pediatric rehabilitation medicine should occur early in the acute period to help maximize recovery (28). As with other children with spinal cord injury, children should receive interdisciplinary, family-centered, goal-directed rehabilitation that focuses on recovery of lost function and making adaptations when function does not return. Depending on the timing of admission to rehabilitation and the capabilities of the rehabilitation unit, some children with transverse myelitis may still be receiving steroids, IVIg, or plasmapheresis for disease management. If a child is transitioned to rehabilitation early in the disease course, vigilance is required to monitor for worsening symptomatology because the time to reach nadir is variable (28). The child with cervical transverse myelitis is at particular risk for decompensation and should be monitored closely for changes in respiratory status due to disease progression. If the lesion is cervical or thoracic, the aggressive pulmonary toilet is recommended. Children with tetraplegia may need cough assist; therefore families should be trained to assist with “quad coughing.” Later in their rehabilitation course, children with transverse myelitis lesions above T6 are at risk for autonomic dysreflexia (see Chapter 16) (28). Chronic sensory changes have been noted in nearly half of patients (32). If bothersome, long-term antiepileptic mediation should be considered. Amitriptyline may also be used in the management of pain (28). A desensitization program may be considered for the child experiencing allodynia or dysesthesia that is interfering with activities. During the acute rehabilitation phase and long term, children with transverse myelitis are at risk for skin breakdown due to limited sensory input and limited mobility. Prevention of skin breakdown is the most effective management and requires keen attention to at risk areas. The patient and family need to be educated extensively and equipment that minimizes risk should be provided. Neurogenic bladder is noted in most cases in the acute period and may persist for a majority of patients (32). Intermittent straight catheterization remains the mainstay of management. Chronically, children may have detrusor muscle spasticity, dyssynergia, or an are-flexive bladder. Urodynamics and routine renal and bladder ultrasound should be a part of the long-term management to ensure urinary system function and health (35). In the very young child with neurogenic bladder associated with transverse myelitis, social continence is not an issue, but catheterizations and medications may still be indicated for urinary system health. For older children, urinary system health and achieving continence should be the goals of treatment. Similarly, neurogenic bowel and the associated constipation often require long-term management (35). Children with spastic bowels usually benefit from an oral agent to improve transit and an agent administered rectally to initiate evacuation. While not immediately evident by the clinical presentation of transverse myelitis, cognitive functioning of the patient with transverse myelitis requires special attention because some patients will go on to develop polyphasic disease. Additionally, in a cohort of children with transverse myelitis, most had neuropsychological function in the normal range but attention and memory problems were identified in a large minority of patients, with a third of parents endorsing subclinical attention problems (36). Neuropsychological screening can identify the need for administration of a full neuropsychological battery. Furthermore, children with transverse myelitis are at elevated risk of depression and anxiety symptoms. Therefore screening and counseling if necessary should be part of the multidisciplinary management team (28). Children deemed at increased risk of progression to polyphasic disease may benefit from baseline neuropsychological testing even in the absence of neurocognitive or neurobehavioral concerns. Because transverse myelitis affects multiple areas of function and can be associated with future polyphasic disease, routine multidisciplinary management is warranted (36). Unlike for children with pediatric MS, where the neurologist is likely the subspecialist with the most active management responsibilities, children with transverse myelitis have needs similar to other children with spinal cord injury, which are often best met by the rehabilitation physician and team. NEUROMYELITIS OPTICA NMO is a polyfocal and often polymodal demyelinating condition characterized by optic neuritis and transverse myelitis (3,33). A relapsing course of NMO, previously known as Devic disease, is more common than a monophasic course (37). NMO used to be considered a rare variant of MS but is now understood to be a separate disease (38). The key to diagnosing NMO includes ensuring that the child’s presentation is not more consistent with MS. This is important because management differs, NMO is less responsive to treatment, and it has a poorer prognosis than MS (38,39). Seropositivity for NMO-IgG is not required for the diagnosis but is highly indicative. Children are seropositive less frequently than adults (3). The binding of NMO-IgG (aquaporin-4-IgG) to aquaporin-4 results in a biological cascade that results in inflammation and demyelination characteristic of the disease (37). The understanding of NMO has expanded exponentially in the past decade and consensus has been reached regarding the diagnosis (40). Table 19.6 details the diagnostic criteria for NMO (41). TABLE 19.6 THE DIAGNOSTIC CRITERIA FOR PEDIATRIC NEUROMYELITIS OPTICA Optic neuritis Acute myelitis Two of the following: • Spinal MRI demonstrating contiguous lesion extending at least three vertebral segments • NMO-IgG seropositivity • Brain MRI does not meet the criteria for MS NMO spectrum disorders are those disorders that do not meet diagnostic criteria for NMO and include recurrent optic neuritis and recurrent transverse myelitis, with seropositivity (6,40). The child with an NMO spectrum disorder may go on to meet the criteria for NMO. Because of this, children with NMO spectrum disorders need to be monitored very closely for disease progression. Children who have relapsing optic neuritis without evidence of other neurologic deficits or systemic disease have chronic relapsing optic neuropathy (CRION), which is often bilateral and steroid dependent (42). These children also have to be closely monitored for progression. Epidemiology of NMO NMO is exceptionally rare; for every individual with NMO, there are 50 to 100 individuals with MS (38). NMO can affect children and adults of any age, but children tend to present in late childhood or adolescence (3). There is a strong female predominance, even stronger than the female predominance for MS (37,39). NMO is more common among those of Asian and African descents (33). NMO tends to occur sporadically but familial cases have been identified (40). A large majority of patients with NMO have serologic evidence of autoimmunity (37). Children with NMO are often seropositive for antinuclear, SS-A, antiacetylcholine receptor, or double stranded DNA antibodies (38). Autoimmune conditions such as SLE, Sjogren syndrome, juvenile idiopathic arthritis, and Graves disease may coexist or precede NMO (37). Clinical Course of NMO and Diagnosis The hallmarks of NMO are transverse myelitis (see preceding section) and optic neuritis. Children with optic neuritis can present with unilateral or bilateral blurry vision, a black spot in the central visual field, or visual acuity loss (43). Compared to adults, vision loss is more severe in children. Children may also report pain with ocular movement (3). If the diagnosis of optic neuritis is suspected, the child should be seen by an ophthalmologist or neuro-ophthalmologist, if one is available. A normal fundoscopic exam does not rule out the diagnosis as some cases are due to retrobulbar inflammation. Visual acuity, visual field, low contrast sensitivity, and color vision should all be assessed. The neuro-ophthalmologist may also perform visual evoked potentials and may recommend optical coherence tomography and an orbital MRI (3). Figure 19.3 demonstrates active optic neuritis. The differential diagnosis list for optic neuritis includes vitamin B12 deficiency, adrenoleukodystrophy, Leber hereditary optic neuropathy (usually pain is absent but a family history is often present), and optic glioma. Additionally, sarcoidosis may present with cranial nerve defects and optic neuritis (3). FIGURE 19.3 This coronal view MRI demonstrates left-sided optic neuritis in a child with NMO. Source: Image courtesy of Guilio Zuccoli, MD, Associate Professor of Radiology, University of Pittsburgh. The treatment of isolated optic neuritis is high-dose steroids (3,44). Refractory cases may be treated with IVIg or plasmapheresis (3). Children tend to recover well from optic neuritis with a majority achieving at least 20/40 vision at follow-up (43,45). Children with bilateral optic neuritis have an increased likelihood of developing MS, as are children who are older (45,46). Children with relapsing optic neuritis may go on to develop either MS or NMO. The case of seropositivity and relapsing optic neuritis without other signs of NMO is NMO spectrum disease. Because children with optic neuritis can later develop MS or NMO, the workup should include a gadolinium-enhanced MRI of the brain and spinal cord and a lumbar puncture with opening pressure (3). Because NMO can occur concurrently with other autoimmune conditions, additional serologies should also be ordered. Similar to other pediatric demyelinating conditions, children often present after a prodromal flu-like illness (37). The child with NMO tends to present with complete transverse myelitis. This is in contrast to MS in which children tend to present with partial lesions (37). Occasionally, though, central cord syndrome may be the presentation of transverse myelitis in NMO (33). A careful and thorough neurologic examination is necessary to delineate the findings of NMO. While transverse myelitis and optic neuritis are the hallmarks of the condition, children with NMO may also have signs and symptoms of intracranial pathology. The MRI of the brain typically demonstrates lesions in the areas of the brain that are aquaporin-4 rich such as the hypothalamus, subcortical white matter, and the periventricular gray matter (3). Brain lesions are more common in pediatric NMO than adult NMO (37). In addition to the typical MRI findings, “cloud-like” enhancements, linear lesions extending from the brainstem to the spinal cord, and posterior reversible encephalopathy syndrome (PRES)-like lesions may be seen (40). Necrosis can occur in the areas of the brain with severe antibody-mediated inflammation and demyelination (37). Children with NMO may also present with encephalopathy and can therefore resemble ADEM. The MRI lesions may be quite large and be radiographically difficult to distinguish from ADEM (3,37,40). In patients with MRI findings in the area postrema, the clinical presentation often includes weeks to months of intractable emesis (37). Treatment of NMO The treatment of NMO includes management of acute episodes, prevention of relapses, symptom management, and rehabilitation (40). The medical management for pediatric NMO is based on the treatment recommendations for adults. First-line treatment is IV methylprednisolone 30 mg/kg/day for 5 days. Plasmapheresis and IVIg are also treatment options in the acute period (37,40). Plasmapheresis is considered a first- or second-line agent and may be given concurrently with steroids or instead of steroids if the child does not tolerate steroid treatment. Five exchanges scheduled every other day is the typical administration and can lower NMO titers (3). IVIg at a total dose of 2 g/kg given over 2 to 5 days is a third-line agent (3). Long-term immunosuppression is the standard of care but is associated with significant side effects about which the patient and parents should be educated (40). Children should be treated with calcium and vitamin D to promote bone health, and they need surveillance because of the risk of bone demineralization (37). For children who are seronegative and fully recovered, prophylactic medications can be considered versus periodic monitoring for new symptoms. Children who are seropositive or who have had a relapse should be placed on prophylactic medication after treatment of the acute episode. Usually oral steroids are given for several months while transitioning to a steroid-sparing agent such as azathioprine, mycophenolate mofetil, or rituximab (3,37). Rituximab dosing tends to follow the schedule developed for rheumatoid arthritis and mitoxantrone, a potent immunosuppressant, has been used in refractory cases (40). Other medication options include methotrexate, IVIg, cyclophosphamide, cyclosporine, and tacrolimus. Plasmapheresis on a routine basis may also be considered (40). It is important to note that many conventional MS medications (interferon-beta, natalizumab, and fingolimod) are contraindicated in NMO because they have been associated with worsening of symptoms (40). Rehabilitation of NMO Unfortunately disability in NMO tends to be more severe than in MS. Disability associated with NMO maps to the severity and location of the lesions with vision loss and paraplegia being the most common (37). Disability tends to accrue with each subsequent attack, so it is important to minimize the impact of each attack and prevent future attacks (40). Pain, stiffness, weakness, fatigue, and bowel and bladder dysfunction are common with NMO. Patients with NMO can experience painful tonic spasms of the limbs that look dystonic in presentation. Carbamazepine has been shown to be effective in reducing these spasms (40). The rehabilitation management of NMO includes the management of transverse myelitis as described in the previous section and in Chapter 16. Additionally, children with brain lesions should receive appropriate brain injury rehabilitation. The rehabilitation program should be interdisciplinary, family-centered, and goal-directed. Neuropsychological testing may be conducted after the acute recovery period to guide classroom adaptations. Like the other polymodal pediatric demyelinating conditions, periodic neuropsychological screening is warranted and repeat testing is often indicated. Unlike the other pediatric demyelinating conditions, except occasionally pediatric MS, low vision rehabilitation may need to be incorporated into the rehabilitation program. Optimally, low vision rehabilitation is a multidisciplinary endeavor that provides assessments, adaptive techniques and strategies, and training (47). Functional vision assessment is the measurement of how well patients use their existing vision to conduct day-to-day activities (47,48). The ophthalmologic assessment should include measurement of visual acuity, contrast sensitivity, perimetry, light characteristics, refractory errors, oculomotor functions, and cortical visual integration (47). Based on the findings, optical assistive devices may be prescribed. Occupational therapists with specialized low vision rehabilitation training teach patients how to use the assistive devices in everyday activities and help patients strategize compensatory strategies (49). Community reentry, including return to school, may be particularly complicated for the child with NMO because of the combination of motor, cognitive, and vision deficits. Environmental assessments and adaptations of the home and school can help improve participation and quality of life by assuring a safe environment for optimal functioning (48,49). While the prognosis remains relatively poor for NMO, advances in the management will hopefully lead to improvements in prognosis in the next several years (40). Actively employing rehabilitation strategies to maximize function in conjunction with immunosuppression and careful multidisciplinary surveillance should improve clinical outcomes and the quality of life of children with NMO. ACUTE DISSEMINATED ENCEPHALOMYELITIS ADEM is a polyfocal demyelinating disease more commonly diagnosed in children than adults (3). To be diagnosed with ADEM, the encephalopathy (behavioral change or altered consciousness) cannot be explained by fever, an infectious process, or a postictal state (6). Additionally, the presentation must be the first demyelinating illness for that child, the MRI must be abnormal in the acute phase (first 3 months), and new findings cannot emerge after the acute phase (6). The typical brain MRI findings in ADEM are diffuse, large white matter lesions with poor demarcation. There may also be focal punctate lesions, bithalamic or basal ganglia lesions, or more rarely, hemorrhagic lesions consistent with hemorrhagic leukoencephalopathy (50). Epidemiology of ADEM The estimated incidence of ADEM is 0.4/100,000/year (51), making it a rare disease. Children commonly present between the ages of 5 and 8 years old and are more often affected than adults. Some cases of ADEM can be linked to preceding viral illness or vaccine administration (Semple rabies vaccine, small pox vaccine, and older measles vaccines), but most are not linked to a specific pathogen (3,52,53). Clinical Course of ADEM and Diagnosis A rapid onset of encephalopathy with multifocal neurologic deficits is the typical presentation with maximal deficits noted between 2 and 5 days after onset (51). A viral infection or vaccine may be in the recent history (2–4 weeks prior) (3,52). Prodromal symptoms can include nausea, vomiting, fever, headache, and general malaise. The level of encephalopathy can range from behavioral changes such as irritability to coma. Children may also present with seizures. Common neurologic signs and symptoms involving the brain and spinal cord include hemiplegia, pyramidal signs, vision changes (due to optic nerve involvement), and speech impairments (3). When ADEM is suspected, a full neurologic examination is warranted. An MRI of the brain and spinal cord should be ordered—lesions may or may not enhance with gadolinium. The MRI images in Figure 19.4 demonstrate multifocal lesions associated with ADEM in a young boy. Rapid determination of a potential viral etiology such as herpes simplex virus (HSV) is essential. Empiric treatment with acyclovir is warranted if a viral etiology is suspected. Cerebral spinal fluid and serologic studies should be collected to help rule out an infectious etiology. Bacterial, viral, and fungal cultures, as well as assays for HSV, Epstein–Barr virus, enterovirus, West Nile virus, and varicella zoster virus (VZV) should be sent (54,55). If anti-NMDAR encephalitis is suspected, then titers should be drawn. Collecting extra cerebrospinal fluid (CSF) for additional studies is wise as it may eliminate the need for a repeat lumbar puncture. The cerebral spinal fluid of ADEM is characterized by normal opening pressure, normal glucose, and moderately elevated cell count and protein (3). Rarely children will experience severe decompensation with increased intracranial pressure warranting surgical intervention (3). Approximately 2% of children with ADEM experience fulminant hemorrhagic demyelination with cerebral edema (50). This condition, called acute hemorrhagic leukoencephalitis is often fatal (55). Treatment for ADEM The first-line treatment for ADEM is corticosteroids intravenously, usually given as 1 g/day for 5 days in children over 40 kg or 20 to 30 mg/kg/day for smaller children. In addition to acyclovir, antibiotic coverage may also be started (3). IVIg is a second-line treatment which can be used in refractory cases or in situations where steroid treatment is not tolerated (3,55). The typical dose of IVIg is 2 g/kg over 2 to 5 days. Plasmapheresis with three to five exchanges may also be used in refractory cases (56). Cyclophosphamide has been used in some cases (55). In the acute period, supportive interventions in the intensive care setting may be necessary to maintain adequate ventilation in cases of respiratory failure, to control blood sugars, and to manage nutrition. After the nadir is reached and the child is stable from a respiratory standpoint, they may be transferred to an acute floor or directly to comprehensive inpatient rehabilitation if it is warranted. FIGURE 19.4 These two axial MRI FLAIR views of the brain demonstrate multiple lesions with increased diffusivity consistent with ADEM. The two largest lesions on each view are indicated with arrows. Source: Images courtesy of Guilio Zuccoli, MD, Associate Professor of Radiology, University of Pittsburgh. Rehabilitation for ADEM The literature on the rehabilitation of ADEM is sparse (61). Children hospitalized with ADEM who have persistent functional deficits should be considered for comprehensive inpatient rehabilitation. Debility may be superimposed upon the neurologic findings of ADEM and children treated with steroids may also have evidence of myopathy. Cognitive and language recovery lags behind motoric recovery, although some children will have lasting physical disabilities (56,58,61). Comprehensive inpatient rehabilitation should focus on recovery of lost function, be it family-centered and goal-directed. Speech therapists, neuropsychologists, and school reentry specialists are particularly important members of the rehabilitation team as many children may have lasting difficulties with attention and executive functioning (62,63). Children with ADEM may have subtle or more overt cognitive challenges and may also present with ongoing behavioral problems. Younger children tend to present and persist with behavioral problems more frequently than older children (3). Children with ADEM may benefit from adaptations in the classroom short term or long term depending on the persistence of their cognitive deficits. Neuropsychological testing may be performed shortly after the acute phase of the disease and should be repeated if abnormalities are identified. PEDIATRIC ENCEPHALITIDES PEDIATRIC ENCEPHALITIS In this section of this chapter, encephalitis in children will be discussed. The first portion will address encephalitis generally. Then specific types of encephalitis will be described in more detail with particular emphasis on autoimmune encephalitis (excluding ADEM, which was described in the “Pediatric Demyelinating Diseases” section). The newly identified condition, anti-NMDAR encephalitis, will be highlighted. Encephalitis is inflammation of the brain tissue with evidence of neurologic dysfunction (54,64). Encephalitis is different than encephalopathy and causes of encephalopathy such as toxic exposure should be ruled out (55,65). Generally speaking, encephalitis may be associated with infections or other inflammatory processes (54). There are well over 100 known pathogens that cause infectious or postinfectious encephalitis (64). There are also numerous noninfectious processes that cause encephalitis. A definitive etiology is identified in only about half of the cases (64). To be diagnosed with encephalitis, a child should exhibit neurologic dysfunction evidenced by altered mental status (behavior or level of consciousness) and have neuroimaging abnormalities consistent with inflammation and/or inflammatory cells in the cerebral spinal fluid (54,64). Epidemiology of Pediatric Encephalitis The estimated incidence of encephalitis has been reported to be 10.5 per 100,000, but is more common in infants (64). Surveillance systems vary internationally and likely underestimate the incidence of encephalitis (54). Additionally, the risks vary between populations. For example, neonates are at much higher risk of HSV encephalitis than other populations. Boys and girls tend to be equally at risk except for autoimmune encephalitis for which girls are at increased risk (54,55). Measles, mumps, and VZV can cause encephalitis (54) and may be on the rise, given the increasing loss of herd immunity associated with the decreased rates of immunization in the United States. Clinical Course of Pediatric Encephalitis and Diagnosis The typical presentation for encephalitis is prodromal “flu-like” illness followed by changes in behavior or consciousness, headaches, and nausea and vomiting. Children may also present with focal neurologic signs and/or seizures (54). Some presentations are more likely to be associated with particular types of encephalitis. For example, children with ADEM often have multifocal neurologic signs, HSV is often characterized by temporal lobe seizures, and arboviruses typically present with fever and early obtundation (64). Important aspects of the history taking at the time of presentation include detailing potential exposures, recent illnesses and vaccines, history of autoimmune disorders, and immunosuppression status (64). In addition to a detailed physical with a complete neurologic examination, neuroimaging and CSF and serum studies are necessary. Blood work should include a culture, CBC with differential, renal, liver, and coagulation studies (54,64). The CSF usually shows lymphocytic pleocytosis, normal glucose, and elevated protein. Oligoclonal IgG bands are rarely seen. Serum and CSF IgM antibodies and/or rising IgG may help the identification of specific viral etiologies (54). CSF viral detection by polymerase chain reaction (PCR) amplification should also be sought to identify specific viral etiologies (54). Samples of both blood and CSF should be stored for future study and a repeat lumbar puncture may be needed when an etiology is not readily identified. Because of the challenges and expense associated with determining a specific etiology, investigations should be directed toward determining if the encephalitis is caused by an autoimmune process versus a viral process to help guide treatment (65). MRI findings vary with etiology. Figure 19.5 demonstrates several small lesions in a child with encephalitis. For the child with suspected seizures, an EEG should be ordered. Nonspecific findings of diffuse high-amplitude slow waves are often seen (54). Although not pathognomonic for HSV encephalitis, temporal lobe periodic lateralizing epileptiform discharges are highly suggestive (54,64). The clinical course is variable depending on the etiology. Some children may require prolonged care in the intensive care setting and may have long-lasting consequences of their disease.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


