Fig. 3.1
Multisystemic symptoms of DM
3.2 Clinical Features
3.2.1 Adult-Onset DM1
3.2.1.1 Muscle Weakness
The predominant symptom of adult-onset DM1 is facial and distal muscle weakness. Facial weakness is an early feature and atrophy of temporalis and masseter leads to the characteristic facial appearance, so-called hatchet face. Mild ptosis is often seen, but ophthalmoplegia is rare. The neck flexors and sternocleidomastoid muscles are commonly involved. Weakness in the limbs is initially distal, especially finger and wrist flexors and ankle dorsiflexors, leading to difficulty with opening caps and foot drop. Muscle weakness progresses gradually and proximal limb weakness occurs later in the disease course, leading to loss of ambulation. Swallowing and respiratory muscles are often involved in later disease stages, with resultant difficulty in swallowing and breathing.
3.2.1.2 Myotonia
Myotonia, defined as delayed muscle relaxation following voluntary contraction or mechanical percussion, is caused by abnormal muscle fiber membrane activity [5]. Grip myotonia, which is difficulty in relaxing the grip, is often observed in adult-onset DM1. Improvement in myotonia by repeated contractions is termed “warm-up phenomenon.” Myotonia can be elicited by percussion of the thenar or tongue. Myotonia can be recorded with electromyography (EMG) as bursts of repetitive muscle fiber discharges, giving a characteristic sound called “dive bomber” or “motorcycle.”
3.2.1.3 Cardiac Involvement
Cardiac involvement is the second most frequent cause of death in DM1 [6]. Conduction disturbance is quite common, such as first-degree atrioventricular (AV) block and QRS widening, sometimes progressing to third-degree AV block. Atrial tachyarrhythmia and fatal ventricular tachyarrhythmia can occur. Regular ECG monitoring and early implantation of a pacemaker or cardiac defibrillator are critical for the prevention of sudden unexpected death. In contrast to cardiac arrhythmia, cardiomyopathy is not common, though some DM1 patients suffer from severe dilated cardiomyopathy and heart failure.
3.2.1.4 CNS Involvement
Central nervous system (CNS) involvement is frequently seen in adult-onset DM1. The characteristic neuropsychiatric features are mild cognitive impairment, inertia, apathy, and reduced perception of disease symptoms. Hypersomnia is extremely common even in the absence of respiratory involvement. Polysomnography shows nonobstructive apneas and central hypoventilation. Brain MRI scan frequently shows diffuse white matter lesions, particularly in the frontal or anterior temporal lobes.
3.2.1.5 Endocrine Involvement
Although overt diabetes is not frequent, hyperinsulinemia and insulin resistance are common in adult-onset DM1. Hypothyroidism, dyslipidemia, and male infertility because of testicular atrophy are also seen.
3.2.1.6 Other Multisystemic Symptoms
In addition to striated muscle, smooth muscle is affected in DM1. Gastrointestinal problems, such as diarrhea and constipation, are particularly frequent and may lead to pseudo-obstruction and megacolon. Cholecystitis and gallstones are also common because of increased tone of the gallbladder sphincter. Cataracts (typically iridescent posterior subcapsular forms) develop in most adult-onset DM1 patients and are sometimes the only symptom of very late-onset mild DM1. Early frontal baldness is often seen in male and even in some female DM1 patients. Increased risks of cancers of endometrium, ovary, thyroid, and colon have been reported [7].
3.2.2 Congenital DM1
Congenital DM1 (CDM) is not merely a severe early form of DM1 but rather a distinct clinical phenotype. CDM patients are severely affected at birth with hypotonia and generalized muscle weakness resulting in difficulty of sucking, swallowing, and breathing. Some CDM patients present symptoms even in utero, such as reduced fetal movements and hydramnios in later pregnancy. Talipes and tented upper lip (so-called carp mouth) are also seen in CDM. Despite neonatal intensive support, the mortality from respiratory failure remains high in affected infants. Surviving infants gradually improve in motor function, but nevertheless show delayed motor and mental development. Children with CDM can achieve independent walking, but develop myotonia and progressive muscle weakness in early adulthood. CDM is almost exclusively maternally inherited, even from mothers with mild forms of DM (see Sect. 3.3.2).
3.2.3 DM2
Although DM1 is common in populations of European and Asian descent, most DM2 patients are of northern and eastern European descent. DM2 patients also present myotonia and muscle weakness; however, the weakness typically affects the proximal muscles including the neck, elbow extension, and hip flexors. Significant muscle pain and fatigue are common in DM2. Cardiac conduction defects, cataracts, and insulin resistance are seen, whereas cognitive manifestation is rare in DM2. In contrast to DM1, a severe congenital form of DM2 has not been reported to date.
3.3 Genetics
3.3.1 Repeat Expansion in DM1 and DM2
Short DNA tandem repeats are observed ubiquitously across the human genome [8]. A small fraction of these repetitive elements are prone to become mutable and highly expanded. Such genomic expansions of simple tandem repeats cause various neurogenetic disorders, including DM. DM1 is caused by an expansion of CTG repeats in the 3′ untranslated region (UTR) of the DMPK gene [2] (Fig. 3.2). The length of CTG repeats is between 50 and several thousand in DM1, whereas the normal repeat number is between 5 and 37. In general, the length of expanded CTG repeats in blood cells correlates with the age of onset in DM1 [9]. DM2 results from a similar expansion of CCTG repeats in the first intron of the CNBP/ZNF9 gene [3] (Fig. 3.2). The expanded CCTG repeat in DM2 is much longer than that in DM1, ranging from 75 to over 11,000. Importantly, unlike that seen in DM1, the size of expanded CCTG repeats does not correlate with the age of onset in DM2.
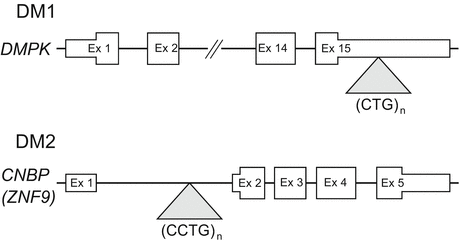
Fig. 3.2
Location of unstable CTG (DM1) or CCTG (DM2) repeats within their respective genes
3.3.2 Repeat Instability
The expanded CTG and CCTG repeats in DM are not stable [10]. These mutations exhibit an exceptional degree of genetic instability in germline and somatic cells. For instance, the size of the repeat is prone to change during transmission from one generation to the next. In fact, intergenerational increments in the order of hundreds of repeats are often seen in DM1 (intergenerational instability). These mutations are also unstable in somatic cells, leading to an age-dependent growth of repeat expansion during the life of an individual (somatic instability).
3.3.2.1 Intergenerational Instability
The expanded CTG repeats in DM1 often increase in size when transmitted from parent to offspring, resulting in earlier onset and more severe clinical symptoms in subsequent generations, a phenomenon called “anticipation.” A maternal expansion bias is evident in DM1 and cases with CDM are almost exclusively maternal in origin. There are a few cases of repeat contraction through paternal transmission [11]. In contrast to DM1, anticipation is less evident, both clinically and genetically, in DM2.
3.3.2.2 Somatic Instability
The expansion process continues throughout life in somatic cells, at rates that are variable between tissues in DM. This can lead to over tenfold variations of expansion length in different tissues of a DM individual [12]. Progenitor alleles that are estimated in leukocytes in adult-onset DM1 are usually between 100 and 800 CTG repeat, whereas adult biopsy and autopsy samples show typically 2,000–6,000 CTG repeats in the muscle or heart. In DM2, the CCTG expansion length is also considerably longer in the muscle than in leukocytes from the same individual [13]. Since the heterogeneity in fetal tissues is much less extensive, somatic instability causing these highly expanded alleles occurs mainly during postnatal life. The functional significance of somatic instability remains unclear; however, the progression of DM may be depending on the growth of the expanded repeat over time.
3.3.2.3 Mechanism of Repeat Instability
Instability of the expanded repeats is associated with DNA replication, repair, or transcription, which instigate separation of double-stranded DNA, thus promoting the formation of extrahelical slipped strand structures that are substrates for error-prone repair (Fig. 3.3a). Recent studies have shown that the effects of transcription on repeat stability are particularly pertinent for DM1 [14]. The predominance of transcription-induced instability in DM is reasonable as the organs most affected, such as the skeletal muscle, heart, and brain, are those with low rates of cell proliferation but the highest levels of DMPK expression. Recent studies also indicate a role for RNA repeats in transcription-induced repeat instability. RNAs comprised of expanded CUG or CCUG repeats bind to the template strand of DNA, then form RNA:DNA hybrids (R-loops) (Fig. 3.3b). The presence of R-loops triggers the formation of extrahelical looped-out structures on the non-template strand and thereby stimulates error-prone repair, leading to further expansion or contraction [15].
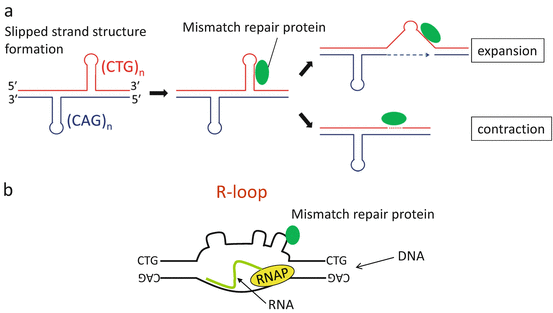
Fig. 3.3
(a) Mechanism of repeat instability. Extrahelical slipped strand structures form in (CTG)•(CAG) repeats, resulting in error-prone processing by mismatch repair proteins. (b) R-loop formation at the DMPK locus. In DM1, expanded CUG RNA binds to the template strand of DNA, then forms RNA:DNA hybrids (R-loops). The presence of R-loops instigates the formation of slipped strand structures on the non-template strand, thereby leading to further repeat instability. RNAP: RNA polymerase
3.3.3 Genotype-Phenotype Correlation
Although the size of expanded CTG repeats in leukocytes correlates with the age of onset in DM1, there was no correlation between muscle weakness and CTG expansion size in either leukocytes or muscle cells [16]. This suggests that some DM1 patients may tolerate longer expansions better than others and raises the possibility that DM1 severity is modulated by additional factors, such as modifier genes, sequence interruptions in the CTG repeat tract, or epigenetic changes at the DMPK locus. The CTG expansion length in the muscle also did not correlate with that in leukocyte from the same subjects [16, 17]. However, the difference in repeat size between leukocytes and muscle is correlated with age, suggesting an age-dependent process of somatic expansion that is more pronounced in the muscle than in leukocytes.
3.4 Pathomechanism
3.4.1 Unstable Repeat Expansion
More than 20 neurogenetic disorders are caused by unstable genomic expansions of simple tandem repeats [8]. Such unstable repeat expansion disorders fall in two categories according to the position of the repeat element within the mutant gene. In most cases the repeat tract is located in protein-coding regions, the motif is CAG, and the orientation in the reading frame produces expanded polyglutamine proteins, resulting in neurotoxicity (e.g., Huntington disease, spinocerebellar ataxia types 1, 2, 3, and 6). The second major group of unstable repeat expansion disorders results from repeats in non-protein-coding regions of genes, such as expanded CGG repeats in the 5′ UTR of FMR1 in fragile X syndrome (FXS) [18], GAA repeats in the first intron of FXN in Friedreich’s ataxia (FRDA) [19], and CTG/CCTG repeats in DM. In cases with FXS and FRDA, the critical effect of the expanded repeats is transcriptional silencing of FMR1 and FXN, respectively. However, there is no proven effect on transcription of DMPK or CNBP/ZNF9 in DM, indicating that the main pathogenic effect is a deleterious gain of function by the mutant mRNA.
3.4.2 RNA Dominance
Although the mutant mRNA is fully processed in DM1, repeat expansion blocks the nuclear export of mRNA with the expanded repeats of CUG (CUGexp) [20]. Thus, the mRNA containing expanded repeats is retained in the nucleus and accumulates in discrete foci. Since DM1 is an autosomal dominant disease, nuclear retention of the mutant mRNA could theoretically lead to a 50 % reduction in DMPK protein. However, several lines of evidence strongly indicate that symptoms of DM1 result not from haploinsufficiency but from the toxic gain of function by RNA transcripts containing the CUGexp. First, DMPK knockout mice, in which DMPK protein can be completely eliminated, show only minor symptoms in the skeletal muscle [21, 22]. Second, muscular features of DM1 can be reproduced by the expression of CUGexp RNA at the 3′ UTR of human skeletal actin transgene in a mouse model [23]. Finally, in DM2, the highly expanded CCUG repeat in CNBP/ZNF9 is fully transcribed, but the intron containing the CCUG expansion is retained and accumulated in nuclear foci similar to that seen in DM1 [3, 24].
3.4.3 RNA-Binding Proteins: MBNL Family and CELF1
The mutant RNA retained in the nucleus affects at least two RNA-binding protein families: the MBNL and CELF. MBNL1, a member of the MBNL family protein, has a strong affinity for CUGexp and CCUGexp [25]. When CUGexp or CCUGexp RNA accumulates in the nucleus, MBNL1 is sequestered in nuclear RNA foci and depleted from the nucleoplasm [26, 27] (Fig. 3.4). Since MBNL1 is a regulator of alternative splicing [28], MBNL1 sequestration in foci leads to the misregulation of alternative splicing of its target exons. One clear example of the functional consequence in DM is the splicing misregulation of CLCN1 [29, 30]. This gene encodes a chloride ion channel that stabilizes the transmembrane potential in skeletal muscle. When alternative splicing of CLCN1 is misregulated in response to expression of CUGexp RNA, the channel activity is lost, causing electrical instability of the membrane, which triggers repetitive action potentials and delay of relaxation (myotonia). The loss of MBNL1 has additional effects on gene expression, including transcriptional alterations, mRNA decay, transport of mRNA, and micro-RNA biogenesis [31–33]. Mbnl1 knockout mice exhibit myotonia and splicing alteration of Clcn1, supporting the evidence for MBNL1 dysfunction in DM1 [34]. Two other members of the MBNL family, MBNL2 and MBNL3, may also be involved in DM [34, 35]. Similar to MBNL1, MBNL2 is expressed in the skeletal muscle, heart, and brain, whereas MBNL3 is expressed mainly in the placenta. MBNL2 and MBNL3 are closely related to MBNL1 and can also be sequestered in nuclear foci. These suggest a role for the combinatorial deficiency of all three MBNL family members in DM, depending on the expression of CUGexp or CCUGexp RNA relative to the distinct levels of MBNL proteins in different tissues. Indeed, a recent report showed compound loss of MBNL1 and MBNL2 functions exaggerated cardiac and skeletal muscle symptoms in a mouse model [36].
Expression of CUGexp also causes perturbations in cell signaling via several kinases, including protein kinase C (PKC), Akt kinase, cyclin-dependent kinase 4 (CDK4), and glycogen synthase kinase 3 beta (GSK3B) [37–39]. While the mechanisms of activation of these pathways have not yet been determined, upregulation of PKC enhances the phosphorylation of CELF1, a member of CELF RNA-binding protein family. The phosphorylation of CELF1 extends its half-life, resulting in the upregulation of its steady-state level [37] (Fig. 3.4). Since CELF1 is a multifunctional RNA-binding protein involved in the regulation of alternative splicing and translation, upregulation of CELF1 protein in DM1 results in misregulated alternative splicing of several pre-mRNAs, as well as altered translation or decay of mRNAs that are bound by CELF1 [40–43]. Transgenic mice overexpressing Celf1 show splicing misregulation and cardiac conduction defect similar to DM1 [44]. As CELF1 plays an important role in developmental splicing switch in the heart, increased CELF1 expression may underlie the cardiac pathology observed in DM1. However, CELF1 upregulation has not been confirmed in DM2 [27, 45].
3.4.4 Spliceopathy in DM
RNA splicing is the mechanism by which intervening introns are removed, while functional exons are ligated together to form a mature mRNA transcript [46]. There are two types of RNA splicing: constitutive and alternative splicing. Alternative splicing serves to generate a wide variety of unique mRNA transcripts by selecting and pairing discrete exons, resulting in distinct but similar proteins from the same gene (Fig. 3.5).
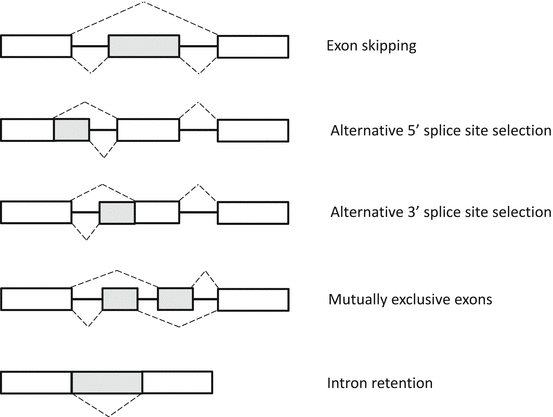
Fig. 3.5
Patterns of alternative splicing. Constitutive exons present in all final mRNAs are indicated by white boxes. Alternative RNA segments that may or may not be included in the final mRNA are indicated by gray boxes
Although many human genetic disorders have been identified as the consequences of mutations on RNA splicing, in almost all cases, these are cis-acting effects that affect the splicing of a single pre-mRNA. In contrast, DM is caused by a trans-effect of alternative splicing factors on many RNAs and now is recognized as the first example of a human genetic disease resulting from “spliceopathy” [4]. The spliceopathy in DM shares common features in splicing misregulation. First, it does not involve constitutive exons, but selectively affects a group of exons that are subject to alternative splicing. Second, it usually affects alternative exons that normally undergo a developmental splicing switch; therefore, the fetal or neonatal splicing isoforms are expressed in mature muscle fibers in DM1. Third, most target exons in the spliceopathy in DM are regulated by MBNL1, CELF1, or both.
More than 70 mis-splicing events have been reported in DM. Table 3.1 lists the transcripts known to be affected by spliceopathy in DM. In case of CLCN1, the exon 7a is negatively regulated by MBNL1 [34]. When MBNL1 is depleted in DM muscle, the exon 7a is retained in the mRNA. The inclusion of exon 7a induces a premature termination codon, which causes loss of mature CLCN1 protein in muscle membrane in DM, resulting in myotonia [29]. Misregulated splicing of insulin receptor (INSR) is also reported in DM muscle [41]. Alternative splicing of exon 11 is an important factor dictating the function of this receptor. The receptor lacking the exon 11 (non-muscle form) has a higher affinity for insulin than the receptor including the exon (muscle form). Because the inclusion of exon 11 is negatively regulated by CELF1, the non-muscle form lacking exon 11 is increased in DM muscle, leading to insulin resistance. However, whereas numerous mis-splicing events have been identified in DM, the underlying cause of progressive muscle wasting, the main disabling symptom in DM, has not been well defined. One possible candidate is mis-splicing of a group of genes regulating Ca2+ homeostasis, such as ryanodine receptor (RYR1), sarcoplasmic/endoplasmic reticulum calcium ATPases (SERCAs), voltage-dependent Ca2+ channel (CACNA1S), and T-tubule membrane scaffolding protein (BIN1) [47–49]. Because Ca2+ concentration is increased in DM1 cells and impaired Ca2+ homeostasis triggers muscle damage, misregulated splicing of these transcripts may cause muscle wasting in DM [50]. Another candidate is splicing misregulation of genes encoding cytoskeletal proteins, such as dystrophin (DMD), dystrobrevin (DTNA), titin (TTN), and ZASP (LDB3), as mutations or deletions in these genes causes other types of muscular dystrophies and myopathies [27, 51, 52].
Table 3.1
Mis-splicing events described in DM
Gene symbol | Function of gene product | Gene symbol | Function of gene product |
|---|---|---|---|
Skeletal muscle | Heart | ||
Cl− ion transport | Cytoskeletal component | ||
CLCN1 | Chloride channel | TNNT2 | Troponin T |
DMD | Dystrophin | ||
Ca 2+ homeostasis | DTNA | Dystrobrevin | |
RYR1 | Ryanodine receptor | TTN | Titin |
ATP2A1 | Calcium ATPase | LDB3 | Z-disk |
ATP2A2 | Calcium ATPase | PDLIM3 | Z-disk |
BIN1 | T-Tubule formation | CAPZB | Assembly of actin filament |
CACNA1S | Calcium channel | ||
Ion channel | |||
Cytoskeletal component | KCNAB1 | Potassium channel | |
DMD | Dystrophin | SCN5A | Sodium channel |
DTNA | Dystrobrevin | ||
TTN | Titin | Others | |
LDB3 | Z-disk | MTMR1 | Phosphatase activity |
CAPZB | Assembly of actin filament | ||
TNNT3 | Troponin T | ||
PDLIM3 | Z-disk | ||
PDLIM7 | Z-disk | ||
FHOD1 | Actin organization | ||
NRAP | Myofibril assembly | ||
ABLIM2 | Z-disk | Brain | |
MYBPC1 | Myosin-binding protein C | APP | Amyloid beta precursor |
MYOM1 | Z-disk | GRIN1 | NMDA receptor |
MAPT | Tau | ||
Signaling | CAMK2D | Signaling | |
INSR | Insulin receptor | SORBS1 | Signaling |
SOS1 | Receptor signaling | DCLK1 | Microtubule polymerization |
ALPK3 | Myogenesis | MPRIP | Rho interacting |
CAMK2B | Signaling | TANC2 | Development |
MAP4K4 | Signaling | KCNMA1 | Potassium channel |
CSNK1D | Casein kinase | ||
Transcription | CACNA1D | Calcium channel | |
MBNL1 | Alternative splicing | LIMCH1 | Actin binding |
MBNL2 | Alternative splicing | ADD1 | Cytoskeletal protein |
NFIX | Transcription factor | PPP1R12A | Myosin phosphatase |
MEF2C | Transcription factor | CLASP2 | CLIP binding |
NCOR2 | Transcriptional silencing | RYR2 | Ryanodine receptor |
FXR1 | mRNA trafficking | ||
Others | |||
GFPT1 | Protein glycosylation | ||
IMPDH2 | Nucleotide biosynthesis | ||
MTMR1 | Phosphatase activity | ||
CAPN3 | Intracellular protease | ||
PHKA1 | Glycogenesis | ||
PHKA2 | Glycogenesis | ||
KIF13A | Endosome positioning | ||
VPS39 | Vesicle trafficking | ||
ARFGAP2 | Vesicle trafficking | ||
COPZ2 | Vesicle trafficking | ||
TBC1D15 | Intracellular trafficking | ||
ANK2 | Membrane targeting | ||
OPA1
Related posts: Toward Regenerative Medicine for Muscular Dystrophies
Therapeutic Approach of iPS Cell Technology for Treating Muscular Dystrophy Toward Regenerative Medicine for Muscular Dystrophies
Therapeutic Approach of iPS Cell Technology for Treating Muscular Dystrophy
 Molecular Pathogenesis and Therapeutic Strategy in GNE Myopathy Molecular Pathogenesis and Therapeutic Strategy in GNE Myopathy
 Stem Cell-Based Therapy for Duchenne Muscular Dystrophy Stem Cell-Based Therapy for Duchenne Muscular Dystrophy
 Translational Research on DMD in Japan Translational Research on DMD in Japan
 Patient Registries for International Harmonized Clinical Development Patient Registries for International Harmonized Clinical Development
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|