Fig. 4.1
RV in GNE myopathy muscle. Bar 20 μm
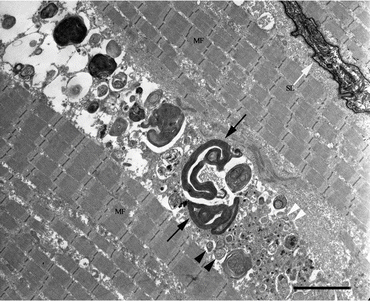
Fig. 4.2
Electron microscopic findings of GNE myopathy muscle. Autophagic vacuoles (black arrow head) are seen in the regions with various cellular debris and protein deposit (white arrow head). Multilamellar bodies (black arrow) are also formed. Myofibrils (MF) themselves are maintained to be almost normal. Sarcolemma (white arrow) is intact. Bar 5 μm
4.4 Genetic Cause and Proposed Molecular Mechanism of GNE Myopathy
More than 100 mutations in the GNE gene have been reported so far [4, 5]. It is known that different mutations lead to different ranges of severity in symptoms [4, 5, 14]. For example, the patients with homozygous p.V572L mutations exhibit earlier onset age and the time from disease onset to wheelchair use is shorter. On the other hand, the patients with p.D176V mutations show later onset age and most of them are ambulatory at the older ages.
GNE gene encodes a bifunctional enzyme working on the rate-limiting step in sialic acid biosynthesis in higher vertebrates. GNE protein catalyzes the epimerization of UDP-GlcNAc to ManNAc and the phosphorylation of ManNAc to ManNAc-6-P. The phosphorylated ManNAc eventually is converted to NeuAc and then CMP-NeuAc. The final product CMP-NeuAc is used as a sialic acid donor for sialylation of gangliosides or sialoglycoproteins in Golgi (Fig. 4.3). Sialic acid production is regulated by a negative feedback inhibition of the GNE activity by CMP-NeuAc. GNE mutations have been reported to lead to decreased GNE activities, subsequently resulting in the reduction in sialic acid production in cells.
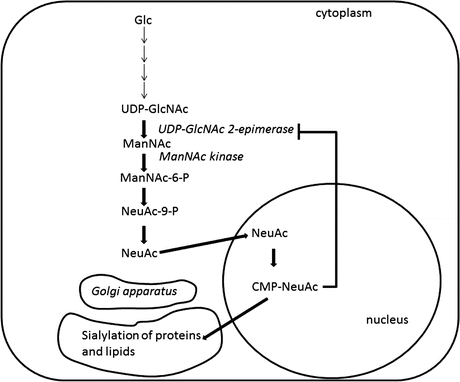
Fig. 4.3
Sialic acid biosynthetic pathway. The initial step in sialic acid biosynthesis is started by a bifunctional enzyme encoded by the GNE gene: UDP-GlcNAc 2-epimerase which epimerizes UDP-GlcNAc into ManNAc and ManNAc kinase which catalyzes the phosphorylation of ManNAc into ManNAc-6-P. The next two steps in the pathway eventually convert ManNAc-6-P into NeuAc. Activation of free NeuAc into CMP-NeuAc occurs in the nucleus. CMP-NeuAc is later transported to the Golgi apparatus and used for sialylation of oligosaccharide chains of gangliosides or sialoglycoproteins in Golgi. CMP-NeuAc regulates UDP-GlcNAc 2-epimerase activity by negative feedback to determine sialic acid biosynthesis in cells
Although decreased sialic acid production in GNE myopathy has been controversial [15], we have demonstrated the hyposialylation in primary culture cells and muscles in GNE myopathy patients [7]. Importantly, the hyposialylation in culture cells was recovered to normal level with NeuAc and ManNAc supplementation [6]. However, no overall hyposialylation in the myoblasts and lymphoblastoid cell line from GNE myopathy patients had been also reported in other papers [16, 17].
4.5 GNE Myopathy Model Animals
Model animals are essential to elucidate the pathomechanism of diseases and to develop therapeutic methods. Three kinds of Gne gene-modified mice and gne-knocked down zebra fish with antisense oligonucleotides have been reported. Two mice strains with the recessive mutations of M712T and V572L (mutation nomenclature is based on GenBank NP_005467) represented the severe glomerular proteinuria without myopathic phenotype [18, 19]. Both mice displayed quite similar renal phenotypes, enlarged glomerulosclerosis, and podocyte foot process abnormalities in the kidney. ManNAc administration to M712T homozygotes and NeuAc administration to V572L homozygotes could prevent the nephrotic-like phenotypes, suggesting hyposialylation would be associated with the phenotypes. The zebra fish treated with antisense morpholino to GNE during embryogenesis showed the impairment of muscle structure, especially reduction in the number of myofibers and large gap between myofibers in electron microscopy [20]. These GNE-mutated/GNE-depleted animals unraveled the new function of GNE proteins; however, these animals must be difficult to be used as the model animals for GNE myopathy for elucidation of myopathy-causing mechanism and development of the therapeutic strategy, since these phenotypes were much different from those in GNE myopathy.
Another model is Gne-knocked out mouse with human GNE transgene with D176V (Gne−/−•hGNED176V-Tg) [21], which is one of the most prevalent GNE mutations among Japanese patients. This model (referred to as “GNE myopathy mice”) represented progressive myopathic phenotype which recaptures those in human GNE myopathy. The experiments conducted on GNE myopathy mice are introduced in this chapter.
4.5.1 Phenotypes of the GNE Myopathy Mouse
The Gne−/−•hGNED176V-Tg mice can be littered in Mendelian rate and no apparent phenotype at birth. The cellular sialic acid levels in various organs other than the brain were remarkably reduced. The weights of the GNE myopathy mice were lighter than control littermates, and this difference in weight becomes more remarkable with age [22]. The motor performance and exercise endurance of the model mice were worse than their littermates, and the impairment of the motor performance became evident from the age of 21 weeks and displayed remarkable disease progression after 31 weeks of age.
In isolated gastrocnemius muscles, peak isometric twitch and maximum tetanic force showed gradual decrements with age: decrease to 90 % by 10 weeks, 80 % by 21 weeks, 70 % by 31 weeks, and 50 % by 41 weeks. The tetanic force was markedly reduced after 41 weeks of age, so the twitch-tetanic ratio is significantly higher in GNE myopathy mice compared with control mice. The gastrocnemius muscles of the GNE myopathy mice were smaller as compared with those of control mice, even in younger age. Furthermore, the muscles demonstrated a steadily decreasing muscle mass of at least 10–20 % from 31 weeks of age and decreasing more remarkably from 41 weeks of age. Cross-sectional area-normalized twitch and tetanic forces showed similar temporal pattern of force reduction in GNE myopathy mice, but interestingly significant differences between GNE myopathy and control mice were only seen from the age of 31 weeks, indicating the only atrophy but not muscle degeneration attributes to muscle weakness in younger age by 30 weeks, and symptomatic muscle degeneration, which would cause the remarkable reduction of tetanic force, begins from 31 weeks of age and results in the severe muscle weakness after 41 weeks.
4.5.2 Atrophic Change Proceeds Development of Pathological Hallmarks in Muscles of GNE Myopathy Mice
No remarkable change in morphological appearance on light microscopy was seen in the muscles from the GNE myopathy mice from 10 to 20 weeks of age, except for minimal variation in fiber size in the gastrocnemius, TA, and QF muscles. The number of small size fibers increased from 21 to 30 weeks of age in gastrocnemius and TA muscles, which contribute to the increasing variation in fiber size. From 31 to 40 weeks of age, a few intracellular inclusions, which were Congo red positive and immunoreactive to various antibodies to amyloid, were observed. The appearance of RVs delayed from 42 weeks (Fig. 4.4). In electron microscopy, the protein inclusions were observed between the bundles of myofibrils in 30 weeks of age, and both the protein inclusions and autophagic vacuoles were accumulated in the center of myofibers apart from the myofibrils after 41 weeks as observed in human GNE patients’ muscles. This accumulation would be results of active cellular response to intracellular inclusion proteins, gathering them into central region by intracellular transport system and inducing macroautophagy process for proteolysis of them.
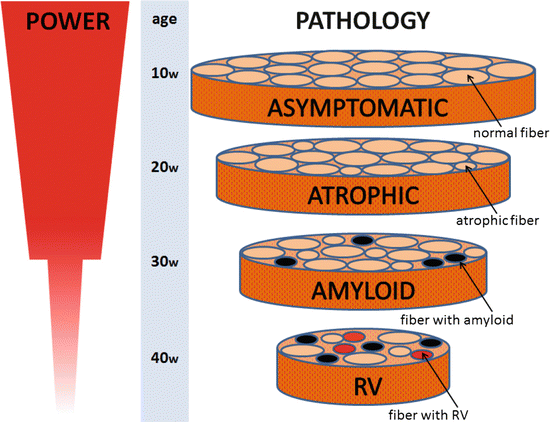
Fig. 4.4
The course of muscle changes in GNE myopathy mouse. No remarkable changes in morphological appearance, but slight weakness in muscle power is seen from 10 to 20 weeks of age. The myofiber atrophy is evident from 21 to 30 weeks of age, which contributes to the increasing variation in fiber size. From 31 to 40 weeks of age, fiber size variation is enhanced and amyloid protein positive – intracellular inclusions are observed. RVs are seen after 40 weeks
Fiber-type selective atrophy was not observed at least in gastrocnemius, TA, and QF muscle. However, surprisingly, almost all fibers that had either RVs or intracellular depositions were type II fibers. In other murine models of muscular dystrophy and myopathies, the predominant involvement of type II muscle fibers [23] has been estimated to be due to the increased susceptibility of these muscles to eccentric contraction-induced damage [24, 25]. In the GNE myopathy mice, this is interesting since the mechanism underlying the disease is remarkably different from these other types of murine myopathy models. Interestingly, in a transgenic mouse overexpressing β-amyloid precursor protein (β-APP), intracellular amyloid deposition has been reported predominantly in type II fibers [26]. This could suggest that the involvement of type II fibers may be secondary to susceptibility to oxidative stress and/or calcium dysregulation attributed to type II fibers. Moreover, in a mouse which overexpresses β-APP in type II fibers, an increase in resting calcium and relative membrane depolarization in muscle fibers have been observed and are thought to represent a mechanism relating β-APP mismetabolism to altered calcium homeostasis and clinical weakness [27]. Because intracellular amyloid depositions are key events in muscle degeneration of GNE myopathy mice, these findings may be of interest for future investigations.
4.6 Treatment of the GNE Myopathy Mouse
We have designed two kinds of the experiments: “prophylactic treatments” that were aimed to prevent onset of the disorder and “amelioration of myopathy phenotypes in symptomatic mice” that was aimed to recover the muscle function from diseased status. The former experiments were also aimed to identify the effective compounds for administration, to clarify the natures of the compounds in animal body, and to get the limitation of the treatments and tolerance of the compounds. The latter experiments were designed as more practical therapeutic applications using weaker animals and a wide range of severity.
4.6.1 Prophylactic Treatment with Sialic Acid Metabolites
ManNAc was effective in preventing the myopathic symptoms of the GNE myopathy mice, equally with low dose (20 mg per kg bodyweight per day), medium dose (200 mg per kg bodyweight per day), and high dose (2000 mg per kg bodyweight per day) [28]. The serum creatine kinase concentration was lowered and motor performance, body weight, and muscle mass were increased after treatment. The number of rimmed vacuoles was significantly lower than the mice who received control treatment. Treatment also increased muscle cross-sectional area and force production and substantially diminished congophilic, amyloid-positive, or tau-positive inclusions. There was no apparent dose effect of ManNAc supplementation in preventing the myopathic symptoms of the GNE myopathy mice. NeuAc and sialyllactose were also effective for the prevention of disease with lower dose at 20 mg per kg bodyweight per day.
To aim for the enhancement of cellular uptake of the administered sialic acid metabolites in mice, we used peracetylated ManNAc for prophylactic treatment of the GNE myopathy mice [29]. Peracetylated ManNAc showed similar recovery of cellular sialylation at less than 1/10 concentration of regular ManNAc in culture medium. As expected, cellular sialic acid levels were highly increased to control levels in all organs including serum, and all aspects in muscle phenotypes, such as muscle power, size, and pathology, were well recovered to those in control mice.
Related posts:
 Toward Regenerative Medicine for Muscular Dystrophies
Therapeutic Approach of iPS Cell Technology for Treating Muscular Dystrophy
Toward Regenerative Medicine for Muscular Dystrophies
Therapeutic Approach of iPS Cell Technology for Treating Muscular Dystrophy
 Stem Cell-Based Therapy for Duchenne Muscular Dystrophy
Stem Cell-Based Therapy for Duchenne Muscular Dystrophy
 α-Dystroglycanopathy
α-Dystroglycanopathy
 Translational Research on DMD in Japan
Translational Research on DMD in Japan
 Patient Registries for International Harmonized Clinical Development
Patient Registries for International Harmonized Clinical Development
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


