CHAPTER 31 Intrathecal Therapies and Totally Implantable Drug Delivery Systems
INTRODUCTION
The discovery of opioid receptors in 19711 and their subsequent isolation in nerve tissue in 19732 in brain tissue3 and the spinal cord were foundational to the idea of using intraspinal opioids for pain control. In 1976, intrathecal opioid infusion was first reported in animals.4,5 Subsequently, intrathecal and epidural morphine was used in humans in 1979.6,7 Both modes of intraspinal delivery were effective in effectively controlling chronic nonmalignant and cancer pain.8 The discovery of a pain processing and modulating role for gamma-aminobutyic acid (GABA) receptors, adrenergic receptors, glutamate receptors, and calcium channels, amongst others, at both spinal and supraspinal levels, expanded intrathecal therapy to include non-opioid agents for intrathecal analgesic application.
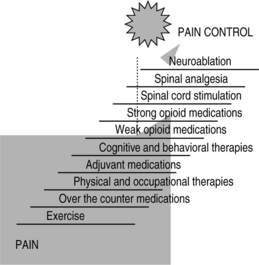
The treatment of chronic nonmalignant pain, like the treatment of cancer pain, requires that the treating physician understands the mechanism underlying the patient’s pain, the psychological and behavioral factors operant in perpetuating that pain, and the appropriate treating modalities to manage the diagnostic specific pain syndrome.9 It is important to know that there are no syndrome-specific therapies and there are many therapies to choose from for the management of chronic pain conditions. Because there are many therapies to treat chronic pain, from noninvasive to more invasive therapies, caregivers should choose therapies using a logical approach or algorithm to those choices. Fig. 31.1 is an example of a pain treatment continuum or algorithm of care that we use in our practice for patients with nonmalignant pain. This continuum of care starts with exercise at the bottom of the ladder moving up, by order of invasiveness and cost, towards more invasive therapies including implantable technologies and neuroablative therapies. This continuum of care should be placed in context by the reader and should not be considered guidelines to care. It is important to emphasize that spinal analgesia is a costly and an invasive therapy and therefore should be applied only when less invasive and less costly therapies have failed, including sequential systemic opioids trials.
INDICATIONS FOR INTRATHECAL THERAPY
Initially, intrathecal therapy was only used for patients with cancer-related pain syndromes, but over the years the indications for intrathecal therapy have expanded.10 Newer indications include neuropathic pain syndromes,11 failed back surgery syndrome (FBSS),12,13 complex regional pain syndromes,14 and head and neck pain.15 Intrathecal therapy is invasive and expensive treatment for chronic pain and should only be applied when certain criteria are applicable. These inclusion criteria for intrathecal therapy include:
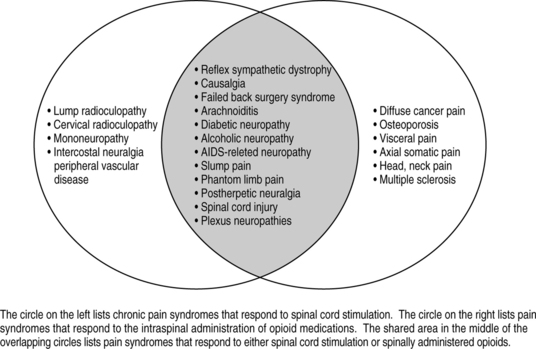
Likewise, when choosing implantable technologies for pain control such as spinal cord stimulation (SCS) or intrathecal therapy with opioids, it is equally important to know if the patient’s pain is nociceptive, neuropathic, or mixed nociceptive/neuropathic. Neuropathic pain is amenable to spinal cord stimulation for pain, while nociceptive pain is not. Intrathecal opioid therapy is useful for nociceptive pain, but minimally useful for patients with primary neuropathic pain. Intrathecal therapy with non-opioids such as local anesthetics, alpha-adrenergic agents or voltage sensitive, N-type calcium channel-blocking agents have been shown to be effective with neuropathic pain syndromes. Because intrathecal therapies are moderately efficacious for neuropathic pain syndromes when using admixtures of opioids and non-opioids there exists no clear-cut boundaries when choosing between SCS or intrathecal analgesic therapies for neuropathic pain syndromes. Fig. 31.2 is a Venn diagram that presents clear-cut diagnoses which respond to intrathecal opioids alone and those that respond to spinal cord stimulation alone. The overlapping gray area represents diagnoses that will respond to both therapies.
TRIALS FOR INTRATHECAL THERAPY
When a patient meets all criteria for intrathecal analgesia delivery and that patient has failed conservative therapies as outlined in Figure 31.1, that patient should undergo a trial for intrathecal therapy.
If the patient does not tolerate the initial drug trialed and is in need of trial of a second agent, only the drug reservoir, the existing tubing and the first 0.22 micron filter which is proximal to the more distal filter (filter nearest the catheter exit site) is changed. Because it is known that infection rates of these systems is proportional to the ‘fiddle-factor’ in personnel handling the systems, we adopt a hands-off policy to system changes and dressing changes. We first change the dressing of the system within 48 hours of surgery and then, unless the dressing becomes wet, we only change the dressing, once every 7 days. We recommend that only physicians or nurses familiar with this system be involved in dressing changes. Biopatch® is left unchanged for 7 days unless saturated with blood.
IMPLANTABLE DRUG DELIVERY SYSTEMS
The first drug delivery system approved for the delivery of intraspinal analgesics was the Infusaid, model #400 pump, which is no longer manufactured. This pump was a nonprogrammable, fixed-rate flow pump. Today, there are other FDA approved fixed-rate pumps including the Codman, model 400 pump (Fig. 31.3) and the Medtronic, Isomed Pump (Fig. 31.4). These pumps utilize a charging fluid that remains a fluid at room temperature, but becomes a gas exerting pressure on a metal bellows that extrudes drug at a fixed rate when implanted into a patient.


Other fixed-rate pumps used outside of the USA includes the Esox®, Archimedes®, Micromedes®, and the Anschutz pump®, all of which are fixed-rate pumps. Some new fixed-rate pumps are being developed by other companies and are not approved by the FDA in the USA today. An example is the AccRx Pump being developed by Advanced Neuromodulation Systems of Plano, Texas (Fig. 31.5). Because the rate of all of these fixed-rate pumps is preset and nonprogrammable, dosing changes are made by changing concentrations of the drug.

Fig. 31.5 AccRx, a fixed-rate pump in trials, manufactured by Advanced Neuromodulation Systems, Plano, Texas.
Medtronic’s Synchromed® system is the only totally implantable and programmable pump that is approved in the USA and Europe. Other companies, including Codman (a Johnson and Johnson company), Advanced Neuromodulation Systems, and Advanced Bionics (a Boston Scientific company), are developing totally programmable intrathecal delivery systems. Rate and therefore dose of drug are externally programmable (Fig. 31.6).

Besides increasing or decreasing a continuous rate of delivery of the drug, the Synchromed® system can be programmed to deliver drug in different configurations that includes programming a single bolus of drug, a timed bolus of drug, a complex continuous delivery of drug, and a continuous delivery of drug.
INTRATHECALLY ADMINISTERED OPIOIDS
Morphine remains the gold standard for intrathecal analgesic therapy. It has an extensive history, with an ample literature for intrathecal therapy, more than any other opioid.16 It is the only opioid approved by the Food and Drug Administration (FDA) for intrathecal analgesia. Other opioids are used for intrathecal therapy when patients are intolerant to intrathecal morphine and include hydromorphone,17 meperidine,18 methadone,19 fentanyl,20 sufentanil,20 and buprenorphine.21 The latter is a partial agonist that is used intrathecally in Europe. These agents are not labeled for intrathecal therapy and are not approved by the FDA, but are used empirically, based on efficacy and safety reported in the literature.
Morphine’s analgesic efficacy is well reported. A prospective survey by Kumar et al. of 16 patients (mean follow-up, 29±12 months) found that intraspinal morphine reduced pain scores for all types of pain by an average of 57%. Surprisingly, the greatest efficacy was found in patients with neuropathic pain and mixed nociceptive/neuropathic pain when compared to nociceptive pain (75% and 61% reductions, respectively).22 A national outcomes registry for low back pain collected prospective data on 136 patients using intraspinal infusion via implanted devices, 81% of whom received morphine. After 12 months, the Oswestry Low Back Pain Disability Scale ratings improved by 47% in patients with back pain and by 31% in patients with leg pain.23
The intrathecal opioid’s time to onset and duration of action, uptake and distribution, availability to supraspinal centers, and central nervous system (CNS) side effects are all governed by the opioid’s relative (to morphine) lipophilicity and its receptor affinity.24 Morphine’s low lipid solubility, high hydrophilicity, and high receptor affinity translates clinically to slow onset of action but prolonged analgesic action. Because of this hydrophilicity of morphine, it remains in the cerebrospinal fluid (CSF) longer and therefore is available to ascend to supraspinal centers through bulk flow of the CSF. Because morphine mixes in the CSF and is carried from low spinal centers to higher spinal centers and supraspinal centers, the position of the catheter tip within the thecal sac is relatively unimportant to provide segmental analgesia. On the other hand, this hydrophilicity of morphine increases the risks for CNS supraspinal side effects such as nausea, vomiting, sedation, and respiratory depression.
Intrathecal morphine delivery is associated with side effects. Both elevated concentrations of morphine-3-glucuronide, a metabolite of morphine in plasma, and an elevated plasma or CSF morphine-3-glucuronide over morphine-6-glucuronide ratio may play a pathogenic role in the development of the cutaneous dysesthesias, hyperalgesia, and allodynia and/or myoclonus seen with the long-term delivery of high-dose intrathecal morphine.25 It seems that morphine-3-glucuronide might exert this neuroexcitatory effect by indirect activation of the N-methyl-D-aspartate (NMDA) receptor, an inotropic glutamate receptor.26 Some patients might not tolerate morphine, but may tolerate hydromorphone, a relatively hydrophilic (to morphine) but more lipophilic agent than morphine. In some patients, supraspinal effects such as nausea and vomiting might be severe. In such cases, more lipophilic compounds which do not mix in the CSF such as fentanyl, sufentanil, or methadone might be chosen.
Intrathecal morphine delivery is known to be associated with hypothalamic–pituitary dysfunction. In a study conducted in Belgium, Abs et al.27 examined hypothalamic–pituitary axis function in 93 patients with non-cancer pain. Seventy-three patients received intrathecal morphine with a mean dose of 4.8 mg/day for a mean duration of 26.6 months. A 20-patient comparison group had comparable pain syndromes but was not treated with intrathecal morphine. A majority of patients of both genders in the intrathecal morphine group developed hypogonadotropic hypogonadism. Fifteen percent also developed central hypocortisolism and/or growth hormone deficiency, compared to none in the control group. Further, decreased libido occurred in 96% of men and 69% of women who received intrathecal opioids, compared with 10% and 20% of men and women, respectively, in the controlled group. Hormone replacement therapy ameliorated decreased libido in 10 of 14 males and 7 of 12 premenopausal females. The authors suggested further investigations to determine the need for systematic endocrine evaluations and replacement therapy in patients treated with long-term intrathecal morphine.
Hydromorphone is a mu agonist that is 8–10 times more lipid soluble28,29 and 5–10 times more potent than morphine. It is a hydrogenated ketone of morphine. Although it is not FDA approved, it is increasingly being used for intrathecal therapy.30,31 Hydromorphone is stable when contained in an infusion system (SynchroMed) and held at 37°C for 4 months.32 Because of its higher lipophilicity than morphine, hydromorphone has a quicker onset of action, a shorter duration of action and, for the same reason, fewer supraspinal side effects because of less rostral spread.
Because of the incomplete cross-tolerance among opioids, switching from morphine to hydromorphone is a consideration when morphine use is associated with too many side effects or poor analgesia. The initial dose of hydromorphone should be 50% of its equianalgesic dose to morphine (equianalgesia is 1:5–10) and should be titrated higher until analgesia is achieved.
Hydromorphone is not only successful in delivering intraspinal analgesia33 but also has a lower incidence of side effects than morphine when given epidurally. In the retrospective study comparing intrathecal morphine to hydromorphone by Anderson et al.,30 37 patients received intrathecal hydromorphone after either inadequate analgesia or unmanageable side effects occurred during treatment with intrathecal morphine sulfate alone. Pain scores improved during the 10 months’ mean follow-up after changing to hydromorphone. Drowsiness and nausea lessened after patients were switched from morphine to hydromorphone and leg edema subsided temporarily, but eventually reoccurred after extended hydromorphone exposure
A known complication of intrathecal therapies is the development of intrathecal granuloma, an intradural but extramedullary mass associated with either high dose or high concentration of intrathecally delivered agents. This complication of intrathecal analgesia will be discussed later in this chapter. In contrast to morphine, hydromorphone exposure intrathecally was not associated with the development of catheter tip granuloma in a sheep model when daily doses, equianalgesic to doses of morphine that produced granulomas, were given.34
However, hydromorphone when used in humans is known to be associated with the development of tip granuloma. In an initial report, the formation of inflammatory masses at the catheter tip was reported in 9 patients who received hydromorphone, either alone or mixed with other drugs.35 A subsequent review of the same report and database and an additional six case reports identified was published with the number of new cases reported in the literature to be 15.36 It still is unclear whether these 15 reported cases of granuloma were due to the intrathecal hydromorphone delivered or due to other agents since all patients had, at one time or another, other agents, either alone or in combination with hydromorphone, in their pumps.
The neuroexcitatory side effects of opioids, e.g., hyperalgesia, are not reported to be more frequent with hydromorphone than morphine, although hydromorphone-3-glucuronide is a more potent neuroexcitant than morphine-3-glucuronide37 which could be responsible for these symptoms.
Meperidine is the only member of the opioid family that has clinically important local anesthetic activity in doses used for analgesia. Because of this quality of meperidine, it is the only opioid in current use that is active as a sole agent for spinal anesthesia (not analgesia). Surgical procedures to the lower limbs, inguinal area, perineum or c-section have been performed using spinal meperidine alone.38,39 Meperidine has been used for labor analgesia and found not to increase c-section deliveries.40 A 0.5 mg/kg intrathecal dose of meperidine produces anesthesia for up to 6 hours or longer.38 A dose of 1 mg/kg or a total dose of 100 mg has been associated with respiratory depression, bradycardia, and hypotension.18,41
There is limited literature on the use of continuous intrathecal meperidine via implantable infusion pumps. In a case report by Harvey et al.42 a woman with chronic low back who failed other medical interventional treatment modalities achieved significant pain relief using a continuous intrathecal meperidine infusion. Another case report also showed similar success.43 According to Chrubasik et al., the dose of meperidine should be 25–30 times higher than morphine to maintain equianalgesia.44 A dose of up to 60 mg per day appears to be safe.45
Methadone. In a survey by Hassenbusch and Portenoy, 2–4% of respondents cited use of this opioid.46 Methadone is more lipid soluble than morphine, hydromorphone, and meperidine, and therefore does not produce noticeable migration within CSF to supraspinal centers. Methadone, based on this lipophilicity, should have fewer supraspinal side effects than morphine when given intrathecally.
The L-isomer of methadone has mu agonist activity and D-isomers have noncompetitive antagonism of NMDA receptors.47 In vitro studies suggest that methadone induces desensitization of the delta opioid receptor by uncoupling the receptor from its underlying G-protein. This delta opioid activity is critical for the development of morphine-induced tolerance and dependence. Animal studies showed that D-methadone reduces the development of morphine tolerance and NMDA-induced hyperalgesia by virtue of its NMDA receptor antagonist activity.48
Studies of the prolonged use of intrathecal methadone for cancer and non-cancer pain showed overall effectiveness between 37.5% and 80% for those populations studied based on greater than 50% reduction49–51 in pain or pain reduction combined with improved scores on quality of life questionnaire.19 These studies involved both cancer and non-cancer patients. Methadone was administered at total daily dosages of 5–60 mg and the duration of treatment ranged from 3 days to 37 months.
Fentanyl is an opioid analgesic which preferentially binds to mu receptors. It is highly lipophilic and has a fast onset of action in the order of 5 minutes with a peak analgesic effect of approximately 20 minutes, when given epidurally. Due to lipophilicity, its effect is largely segmental, although continuous intrathecal infusion is associated with receptor saturation and mixing in the CSF. It is equipotent when given epidurally or intravenously.52
As fentanyl is 75–100 times more potent than morphine sulfate, lower doses are needed to produce similar analgesia. Thus, considerations for side effects must be made, especially since fentanyl side effects have been noted to be more severe than those of morphine sulfate. Cephalad spread of fentanyl which could lead to respiratory depression does occur, but less so than morphine. Although it is uncertain whether fentanyl’s exact location of action is systemic or spinal, the systemic mode of action appears to be favored by some.53 While scientific data on epidural and intrathecal fentanyl are evolving, there does appear to be a role for fentanyl in treating both acute and chronic pain processes.
Two retrospective studies on intrathecal fentanyl have been reviewed. One study of 122 patients examined the complications associated with implantable drug delivery systems. This study included two patients with the combination of fentanyl and bupivacaine54 and neither of these patients experienced serious adverse events. In another study, eight patients out of a total of 29 patients with intrathecal therapy received intrathecal fentanyl, 10.5–115 mcg/day for a mean duration of 31 months.55 The authors reported a 68% reduction in pain and an overall satisfaction of 3.25 on a scale of 1 (poor) to 4 (excellent) in all eight patients.
Due to being extremely lipid soluble, it largely diffuses into the central neural tissue when administered intrathecally, leaving less bulk drug available for rostral movement, and thus fewer central side effects such as nausea, vomiting, itching, urinary retention, and respiratory depression. Because of this lipid solubility, after intrathecal administration, sufentanil concentrations in the CSF decrease more quickly when compared to morphine. The mean residence time in the CSF, i.e., the time required to eliminate 63.2% of the drug, is approximately 0.9 hours after injection of 15 mcg sufentanil56 while that of morphine (0.05 mg/kg) is 2.3 hours.57
The higher affinity of sufentanil to mu opioid receptors when compared to morphine gives sufentanil a potential benefit in delaying the development of tolerance to the drug. It has been postulated that agents with high efficacy and receptor reserve, i.e. sufentanil, should produce less tolerance than agents of lower efficacy and lower receptor reserve, i.e. morphine.58 Rats develop, over time, less tolerance to intrathecal sufentanil when compared to morphine. Also, sufentanil requires the occupancy of fewer mu receptors to produce antinociception than does morphine.
In a survey performed by Hassenbusch and Portenoy,46 nearly 20% of pain clinicians have used either fentanyl or sufentanil in intrathecal drug delivery systems; however, there are no clinical data of long-term use of intrathecal sufentanil in humans. It is known that in humans a 10 mcg bolus of intrathecal sufentanil produces analgesia within 5 minutes with a duration of about 19 minutes.59
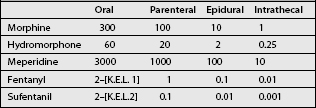
Because of its high lipophilicity, this agent is used when more hydrophilic drugs produce excessive supraspinal side effects. When converting morphine to sufentanil in an implanted pump, we, based on the potency ratio of sufentanil to morphine, arbitrarily use a dose conversion of 1 mcg of sufentanil to 1000 mcg (that is 1 mg of morphine) as shown in Table 31.1
Animal toxicology studies of intrathecal sufentanil have shown no detectable pathological effects on spinal cord histology. However, in a study performed in sheep, which have relatively smaller intrathecal spaces, a rather large dose of 7.5 mcg/kg of sufentanil did produce demonstrable neurotoxic changes. These authors did demonstrate a low-grade inflammatory response to the intrathecal catheter itself and concluded that the response was thought to represent a foreign body response.60
INTRATHECALLY ADMINISTERED NON-OPIOIDS (ADJUVANTS)
Bupivacaine. Local anesthetics are the most common adjuvant agents used with opioids for intrathecal therapy. By the year 2000, more then 20% of patients with intrathecal therapy for pain control had local anesthetics in their implantable pumps. According to this survey of implanting physicians, 60% of the responding physicians used morphine–bupivacaine combinations.46
Bupivacaine HCl has been shown to be stable and compatible with the Medtronic SynchroMed implantable pump, maintaining 96% of its initial concentration with chronic exposure to the system after 12 weeks at 37°C.61 Further, Trissel et al.62 in a stability study, showed that combinations of morphine sulfate, 5 mg/mL plus bupivacaine, 2.5 mg/mL, or morphine sulfate, 50 mg/mL plus bupivacaine, 25 mg/mL, packaged in 20 mL aliquots in plastic syringes and maintained at 4°C or 23°C for 60 days or −20°C or 37°C for 2 days remained stable. In other words, the morphine remained stable at, or greater than, 97% and the potency of bupivacaine remained greater than 95%, after the study. Frozen samples exhibited microparticulates upon thawing, suggesting that freezing of morphine–bupivacaine combinations should be avoided.
Bupivacaine is the intrathecal local anesthetic most widely used in humans. Other local anesthetics including tetracaine and lidocaine have been tried but their use has been abandoned due to their relative known toxicity.63 Recently, ropivacaine has been tried in humans;64 however, it offers little advantage over the less expensive bupivacaine.
Human postmortem studies and the long-term usage of bupivacaine do suggest clinical safety of the long-term infusion of low doses of intrathecal bupivacaine.65 There are, however, reports in the anesthesia literature of permanent neurological sequelae, specifically the cauda equina syndrome, when some local anesthetics, specifically 5% lidocaine, are used for spinal anesthesia, particularly when delivered through small-bore intrathecal catheters.66 It is unclear, as reported by the authors, whether this complication was drug related, catheter related, technique related, or a combination of these.
The positive analgesic response with the use of local anesthetic–opioid combinations intraspinally, for the treatment of cancer and non-cancer-related pain, has been previously reported.67,68 Deer et al., in a retrospective study of 109 patients, 25 with cancer pain and 84 with nonmalignant pain, studied the effect of the addition of bupivacaine intrathecally to patients who did not receive analgesia with their intrathecal opioids alone (morphine, mean dose of 8 mg/day, or hydromorphone, mean dose 1.5 mg/day).69 All patients who had a poor response to the opioid alone, who were switched to opioid–bupivacaine combinations, reported significantly lower pain scores and experienced a mean opioid dose reduction of 23. In a nonrandomized, noncontrolled prospective study, performed by van Dongen et al.,70 it was found that in 5 of 20 cancer patients with inadequate analgesia with morphine alone, the addition of bupivacaine (5–21.6 mg) improved pain relief. However, in the study, the doses of morphine were relatively low (1.2–7.2 mg/day). Recently, Mironer et al., in a randomized, double-blind, multiple-phase, crossover trial, comparing intrathecal morphine or hydromorphone alone to morphine and or hydromorphone with the addition of bupivacaine, found a statistically significant improvement in quality of life scores, but no change in pain scores, in the group with the addition of bupivacaine, when compared to the group with opioids alone.71 Lundborg et al. reported on their clinical experience using intrathecal bupivacaine infusion for three patients with complex regional pain syndrome.72 These 3 patients received bupivacaine at an average dose of 54 mg/day, maximum of 90 mg/day, and were followed for an average of 374 days during their infusion for an average of 1900 days. Although there was a moderate decrease in pain, there was no improvement in allodynia, edema, or trophic changes.
Clonidine is not approved for clinical use in the USA; however, its use is widespread and safety has been established. Because of this, and because of a significant literature on its use, this agent for intrathecal use should not be considered experimental. Some authors have found that the combination of clonidine with an opioid works synergistically and thus the combination provides better analgesia at lower doses than if the drugs were given alone.73,74
Daily intrathecal dosages in humans have ranged from 3 mcg/kg to 60 mcg/kg. Studies in different animals and humans have demonstrated no neuropathology after intraspinal clonidine.75 The main side effect of clonidine, when given intrathecally, is hypotension. Humans may also experience decreases in heart rate but no metabolic changes. Other side effects of intrathecal clonidine include dry mouth, drowsiness, dizziness, and constipation. Sudden withdrawal of clonidine can precipitate agitation and rebound hypertension.76
Some authors believe that clonidine may be useful in facilitating a ‘spinal opioid holiday’1 in patients who have become tolerant to high doses of opioids.77 Clonidine may also be effective in neuropathic pain78 and sympathetically mediated pain syndromes.79
Clonidine has been used safely in numerous patients and is stable with various agents. It is stable in a titanium reservoir infusion pump (SynchroMed, Medtronic, Inc., Minneapolis, Minn., USA) in concentrations of 0.05 mg/mL and 1.84 mg/mL, in combination with morphine sulfate at a concentration of 2 mg/mL and 20 mg/mL at 37°C for 90 days and 54 weeks, respectively.80 When an admixture of morphine sulfate, bupivacaine hydrochloride, and clonidine hydrochloride was incubated in SynchroMed-EL pumps (Medtronic, Minneapolis, Minnesota, USA) at 37°C for 90 days or stored in glass vials at 4°C and at 37°C, as controls, the concentrations remained at greater than 96% of the original concentrations.81 A color change from colorless to light yellow between days 30 and 60 did not affect the solution’s stability. No particulate matter and no clinically significant changes in osmolality were observed during this 90-day study period.
Hassenbusch et al. reported a 20-month prospective phase I/II cohort study of 31 patients (6 with cancer pain, 25 with nonmalignant pain) who received intrathecal clonidine alone at dosages between 144 and 1200 mcg (mean 872 mcg).82 Twenty-two patients achieved greater than 50% pain and symptom reduction without intolerable side effects. At 6 months, 77.3% (17/22) achieved continued good pain relief, and 59% of the total group were considered long-term successes (mean follow-up 16.7 months). Clonidine dosages did not change significantly over time in this group. Poor long-term outcomes were related to inadequate pain relief (n=4), intolerable side effects such as hypotension (n=2), impotence, lethargy, and malaise (n=1 each).
A retrospective survey of Ackerman et al. observed that only 2 of 10 patients treated with clonidine alone (doses 75–950 mcg/day) had good pain relief after 7–11 months of intrathecal therapy.83 The addition of clonidine (75–950 mcg/day) to either morphine 0.15–15 mg/day (5 patients) or hydromorphone 200–800 mcg/day (10 patients) similarly yielded very limited effect; only 3 of these patients achieved long-term pain relief (7–11 months). Although the results from this study and that of Hassenbusch et al.82 appear to contradict each other, it should be noted that Ackerman and colleagues’ report was retrospective in nature and their group was not homogeneous, since many patients received either many drugs over time and/or more than one drug at a time during the therapy period.
Siddall et al., in a randomized, prospective study, compared the efficacy of intrathecal administration of saline, morphine (0.2–1 mg), clonidine (50–100 μg), and the combination of clonidine and morphine.84 In phase I of the study, each patient received saline, clonidine, and morphine in a random sequence. One dose per day of each drug was titrated over 3 days until the occurrence of either analgesia (defined as a >50% reduction from baseline pain score) or side effects. During phase II of the study, each patient received a combination consisting of 50% of the final dosage of morphine combined with 50% of the final dosage of clonidine. The authors compared the proportion of those patients who had a positive response at any time during the assessment. Five of 15 patients responded positively to saline, 3 of 15 responded to the largest dosage of clonidine alone, 5 of 15 responded to the largest dosage of morphine alone, and 7 of 15 to the combination of one-half the largest dosage of clonidine plus one-half the largest dosage of morphine. These data suggest that morphine and clonidine is a worthwhile combination to achieve improvement in analgesia.
In a prospective study, Uhle et al. reported on 10 patients with neuropathic pain syndromes who received clonidine at an average of 44 mcg/day in combination with morphine sulfate or buprenorphine.85 All patients had a 70–100% reduction in pain after the addition of clonidine to either morphine or buprenorphine. Four of eight patients with non-neuropathic pain syndrome also appeared to benefit from the addition of clonidine. The most frequent adverse effects noted in this study were hypotension, fatigue, dry mouth, and impaired bowel (N=10, 4, 3, 1 patients, respectively).
Tizanidine is an alpha2 agonist used clinically as a muscle relaxant. It appears to have a potential role as an intrathecal analgesic drug. In a dog model, Kroin et al.86 reported that tizanidine and clonidine at dosages of 3.0–18.0 mg/day yielded equivalent analgesia using a thermal withdrawal test, but that clonidine, when compared to tizanidine, was associated with greater toxicity (hypotension, bradycardia, and bradyarrhythmias). A further toxicity study of 3–6 mg/day in dogs was performed and these authors found no significant side effects or differences in spinal cord histopathology between the 3 mg/day and 6 mg/day groups.
Kawamata et al.87 studied the effects of clonidine and tizanidine on a rat model of neuropathic pain. Sprague-Dawley rats were chronically implanted with lumbar intrathecal catheters, and the sciatic nerve was loosely ligated. Twenty-one to 28 days after surgery, the rats received intrathecal clonidine (0.3, 1.0, and 3.0 μg) and tizanidine (1.0, 2.0, and 5.0 μg), and the antihyperalgesic effects of thermal and mechanical stimuli were examined. In addition, changes in blood pressure and heart rate, sedation level, and other side effects after intrathecal administration of drugs were studied. The authors found that the administration of 3.0 micrograms of intrathecal clonidine or 5 micrograms of tizanidine significantly reversed both thermal and mechanical hyperalgesia. The administration of 3.0 micrograms of intrathecal clonidine, but not 5.0 micrograms of tizanidine, significantly decreased mean blood pressure and heart rate and produced urinary voiding. A greater sedative effect was produced by the clonidine when compared to the tizanidine. The authors concluded that the antihyperalgesic dose of intrathecal clonidine and the antinociceptive doses produced several side effects. Intrathecal tizanidine at the dose that reversed hyperalgesia would be preferable for neuropathic pain management because of absence of hypotension and bradycardia and lower incidence of sedation.
Ziconotide is an omega-conopeptide, a synthetic form of the cone snail peptide, varpi-conotoxin MVIIA. Ziconotide is a neuron-specific, N-type voltage-gated calcium channel blocking agent with an analgesic and neuroprotective effect.88 Spinally administered ziconotide blocks neurotransmitter release from primary nociceptive afferents to prevent pain signal propagation to the brain. It has an advantage over intrathecal morphine in that it is a non-opioid and tolerance does not developed after its prolonged use. Although it has been used extensively in humans, ziconotide is approved by the FDA for clinical intrathecal use.
Ziconotide is indicated for both nociceptive and neuropathic type of pain. It has been shown to be effective not only for chronic pain but also for acute postoperative pain. Atanassoff et al.89 performed a randomized, double-blind pilot study in patients undergoing elective total abdominal hysterectomy, radical prostatectomy, or total hip replacement. After intrathecal injection of local anesthetic and before surgical incision, a continuous intrathecal infusion of either placebo or 1 of 2 doses of ziconotide (0.7 mcg/hour or 7 mcg/hour) was started and continued for 48–72 hours postoperatively. Thirty patients received the study drug and 26 of them were evaluable for efficacy. It was found that the mean daily patient-controlled analgesia (PCA) morphine equivalent consumption was less in patients receiving ziconotide than in placebo-treated patients. The visual analogue scale of pain intensity (VASPI) scores during the first 8 hours postoperatively were remarkably lower in ziconotide-treated than in placebo-treated patients. In 4 of 6 patients receiving the high dose of ziconotide (7 mcg/hour), adverse events such as dizziness, blurred vision, nystagmus, and sedation led to discontinuation of the study drug infusion. After ziconotide discontinuation, these symptoms resolved.
In a double-blind, placebo-controlled, short-term trial of ziconotide in 257 patients with non-cancer pain by Presley et al.,90 31% of the ziconotide-treated patients reported significantly lower pain scores, compared to 6% of the placebo-treated patients. Moderate to complete pain relief was achieved in 43% of patients in the ziconotide arm versus 18% in the placebo arm. Patients who received ziconotide also decreased their systemic opioid intake and reported improved quality of life compared to patients who received placebo.
Staats et al., in a double-blind, placebo-controlled, randomized trial,91 studied 111 patients (ages between 24 to 85 years) with cancer or AIDS pain with VASPI score of 50 mm or greater. Patients were randomly assigned in a 2 to 1 ratio to receive ziconotide or placebo treatment. Intrathecal ziconotide was titrated over 5–6 days followed by a 5-day maintenance phase for responders and crossover of nonresponders to the opposite treatment group. Pain relief was moderate to complete in 53% of patients in the ziconotide group compared to 17.5% in the placebo group. Five of 111 (4.5%) patients receiving ziconotide achieved complete pain relief.
In spite of being a promising analgesic, ziconotide, like all analgesics, is not free of side effects, and side effects increase with a rapid titration of the agent and decrease with reduction in dose.91 Adverse events associated with intrathecal ziconotide have included vestibular disorders such as nystagmus, abnormal gait, nausea/vomiting and dizziness, urinary retention, blurred vision, diplopia, memory impairment, and orthostatic hypotension.90–92 Apparently, these side effects do decrease over time. In the study by Penn and Paice93 involving the use of intrathecal ziconotide in three patients with both neuropathic and nociceptive pain mechanisms, in dosages between 0.2 mcg/hour and 5.3 mcg/hour, significant pain relief was achieved. Side effects seen included nausea, diarrhea, nystagmus, dysmetria, sedation, confusion, and hallucinations. With intrathecal ziconotide doses of 0.9 mcg/hour or greater, much more serious side effects occurred including disorientation, agitation, and in two of three patients, unresponsiveness, which only resolved at some point after intrathecal ziconotide was discontinued.
Midazolam has been used as a sole drug for intrathecal therapy for the management of postoperative pain.94 In animal studies, it has been shown that the antinociceptive analgesic effect of intrathecal midazolam can be reversed by naloxone; more specifically, this analgesic effect of intrathecal midazolam was blocked by delta-selective antagonists in rats.95 Midazolam has been shown to have a synergistic analgesic effect when given with bupivacaine intrathecally, and in animals this synergistic analgesic effect was demonstrated, giving midazolam with clonidine and with NMDA and AMPA receptors antagonists in rats.96,97
Because of conflicting reports of different toxicities in differing animal models, the use of midazolam, although used emperically by some in humans, continues to be controversial. Toxicity has not been associated with the intrathecal infusion of midazolam in studies on rats,96 cats,98 pigs and sheep;99 however, there has been, in contrast to these studies in rats, cats, pigs and sheep, evidenced toxicity with the intrathecal infusion of midazolam in rabbits.100 For a detailed review of preclinical safety issues with midazolam see the excellent review by Yaksh and Allen.101
In a prospective study by Rainov et al., 26 patients received a combination of various intrathecal agents as follows: four patients received midazolam 0.4 mg/day plus morphine sulfate 0.5 mg/day, clonidine 0.03 mg/day, and bupivacaine 1.0 mg/day; four patients received morphine/bupivacaine/midazolam; and two patients received morphine/midazolam.102 Overall, 19/26 patients (73%) achieved good to excellent pain relief, 6/26 patients (23%) achieved sufficient pain relief, and one patient reported poor pain relief. No information was provided regarding outcomes according to drug combination received on-study.
Baclofen is a GABA-beta agonist that has been used to treat spasticity since 1984. Intrathecal baclofen is FDA approved for the treatment of spasticity caused by upper motor neuron disease; however, questions remain as to whether baclofen is an analgesic when given intrathecally. Baclofen is stable alone or in combinations with clonidine when placed into intrathecal pump systems.103
Other non-opioid agents used intrathecally
Other agents shown to be effective for the relief of pain when given intrathecally include neostigmine,104,105 adenosine,106,107 and octreotide.108,109
Neostigmine is an acetylcholinesterase (Ach) inhibitor which increases the availability of acetylcholine at the neuromuscular and the dorsal horn level. Since muscarinic receptor2 agonists produce antinociception in rats,110 and because muscarinic receptors have been detected at the level of the substantia gelatinosa and to a lesser extent in laminae III and V of the dorsal gray matter of the spinal cord, it was felt that neostigmine would be an effective analgesic if given intraspinally in humans. Neostigmine does produce analgesia in humans; however, its use is associated with an extremely high incidence of nausea and vomiting.105
Endogenous adenosine may be involved in the mediation of the spinal antinociception induced by descending adrenergic fibers originating from the locus ceruleus. This antinociceptive effect is blocked by intrathecal aminophylline (an adenosine receptor antagonist).111 Kekesi et al.106 reported that adenosine has little antinociceptive efficacy during continuous intrathecal administration, but appears to potentiate the effect of endomorphin-1. Eisenach et al.107 compared intrathecal adenosine with intravenous adenosine for chronic neuropathic pain in seven patients with hyperalgesia and allodynia. In intrathecal group, spontaneous pain was not relieved; however, evoked dysesthesias such as allodynia and mechanical hyperalgesia were markedly reduced. Five of seven patients developed back pain after the induction of adenosine.
Octreotide (Sandostatin), a synthetic octapeptide derivative of somatostatin, has been found to be beneficial in the treatment of chronic pain, although the mechanisms underlying its therapeutic effect are not completely understood. Somatostatin is distributed in the substantia gelatinosa. It has been shown to have analgesic effect without adverse effects if given intrathecally, but the high cost, more than US$20 000.00 per year, prevents its widespread use.113 In a recent double-blinded study, octreotide was found to exert primarily an antihyperalgesic rather than analgesic effect on visceral pain perception.112






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


