Fig. 1
Types of biomarkers that can be used at different stages of rheumatoid arthritis
The majority of biomarkers in RA are also molecular like autoantibodies, cytokines, and acute-phase reactants. Genetic markers have long been recognized as markers of susceptibility, clinical classification, and prognostic classification, but none has yet achieved a biomarker status. Recent genome-wide association studies (GWAS) have identified strong markers for susceptibility, disease classification, drug response, and prognosis, but their development into biomarkers will pose difficulties as will be explained below. This is an important issue for RA is one of the diseases with a high heritability [19–22]. The markers identified to date, however, do not account for the large portion of this genetic risk. It is expected that with the expansion of search for genetic markers for predisposition to epigenetic markers [23–25], the “missing heritability” may be uncovered.
Risk Markers Are Not Necessarily Biomarkers
A common misconception is that any marker that shows an association with any aspects of a disease, i.e., a risk factor, can be used as a biomarker. In practice, however, it is not that straightforward [15, 16, 26, 27]. Even a risk factor whose association with a trait yields a relative risk or odds ratio (OR) of 5.0, which is a high value for any epidemiologic association, is unlikely to be an informative biomarker at the individual level. For a risk factor to have a detection rate of 80 % for a false-positive rate of 5 %, the OR should reach more than 2,000, which corresponds to almost exclusive presence in the group of interest. This is exactly the problem with most risk factors, in particular, for genetic markers in that a lot of healthy people will also carry the marker without any sign of the disease. By using the copresence of multiple markers, a threshold may be obtained that the combination of markers would only be present in patients and absent in healthy subjects. Although achievable, this situation will only apply to a minority of patients and a minority of controls resulting in low specificity and sensitivity for the marker as a biomarker.
It may appear paradoxical that a strong risk marker yielding a very high relative risk with a very strong statistical result is not as useful for biomarker development as expected. First of all, a statistical result only shows that the marker is a classifier – in the sense that it classifies a subject as a case or control – and this is less likely to be due to chance. As will be discussed, a likelihood ratio (which incorporates specificity and sensitivity), area under the receiver operating characteristic (ROC) curve analysis, and reclassification statistics are better indicators of the value of a test as a biomarker. The ultimate aim is to have a marker able to classify a subject as diseased or healthy, rather than estimating the risk of disease, i.e., how many folds increased risk a subject has. An ideal biomarker is present in all diseased subjects (100 % sensitivity), and it is totally absent in non-diseased (healthy or those patients with other diseases) subjects (100 % specificity) yielding a likelihood ratio of infinity. As an example, the FDA-approved 70-gene signature in breast cancer for overall survival [28] corresponds to P < 0.001 and a hazard ratio of >2.0, but a sensitivity of 90 % and a specificity of 40 %, with the resulting AUC value of around 0.65 for survival. These figures correspond to a poor-to-modest classifier: to identify correctly 90 of the 100 patients with poor prognosis (and missing 10 of them), 60 of 100 patients with good prognosis are also identified as candidates for poor prognosis [27].
Biomarker Development
The initial discovery phase is just a start in a rather involved process of biomarker development. This process involves (1) discovery and replication, (2) analytic validation, (3) clinical validation, (4) determination of clinical utility, and (5) clinical use following regulatory approvals (Fig. 2). Taking a simple association study to a clinically useful biomarker is a process as detailed and elaborate as drug development [29, 30]. Such a successful translation requires work in basic, translational, and regulatory science and a comprehensive collaboration among laboratory scientists, technology developers, clinicians, statisticians, and bioinformaticians. Several guidelines have been published for different aspects of biomarker development studies: molecular epidemiology (STROBE-ME) [11], early clinical trials of novel agents [31], tumor marker prognostic studies (REMARK) [32], genomic applications (EGAPP) [33], genetic risk prediction (GRIPS) [34], and biospecimen-based studies (BRISQ) [35]. These and other [36–38] guidelines aim to prevent the use of biomarkers in the absence of high levels of evidence supporting their clinical utility and help the investigators to design biomarkers that can lead to true personalized care with high confidence.
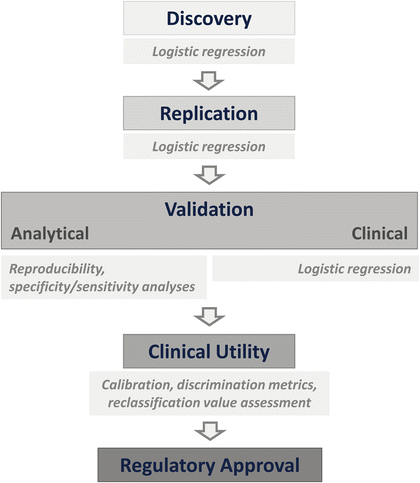
Fig. 2
Steps to be taken to convert an experimental finding into a clinical biomarker
It is important to appreciate that the discovery phase is just the beginning, but still strict guidelines of study design should be followed [30, 39, 40].
The discovery phase studies tend to be retrospective and small in size and suffer from well-recognized problems such as cross-validation for replication and extensive subgroup analysis. Instead, emphasis should be given to statistically adequately powered, prospective studies. This can be achieved using stored biospecimens of ongoing or completed cohort studies or randomized clinical trials as long as samples from placebo arm or standard treatment are used [30], bearing in mind the caveats of inclusion criteria for clinical trials. Since the rest of the development program will be based on these initial results, extreme care at this stage will pay off later.
Once the initial results are obtained in the discovery phase, the validation phase begins with examination of analytical validity as the next step. This is a technical checklist of the analysis methods used in the measurement of the biomarker candidate. The specific assay test for the biomarker is examined for its accuracy and precision. For a genetic variant, this step makes sure that the genotyping assays perform well with acceptable concordance rates by different operators, in different laboratories using different platforms. Analytical validity is measured in the laboratory by calculating the test sensitivity, which provides the probability that a positive test is truly positive (e.g., will yield is a positive results when the marker is in fact present), and test specificity, which provides the probability that the test will not detect the marker when it is not present. This calculation is performed by testing the analysis method against a gold standard or by using samples known to be positive and negative for the presence of the marker. Ideally, both parameters should be 100 %.
In the clinical validity step, the power of the biomarker candidate to show statistical correlation with the phenotype of interest or intended clinical endpoint is reexamined and confirmed. It is in this step that potential confounders (age, sex, race/ethnicity, die, medications) are also examined. Measures of sensitivity and specificity are evaluated in a representative sample of the population for whom the test is intended using appropriate epidemiologic study designs ideally including an independent replication study. These studies also establish the positive predictive value, which reflects the probability that a person with a positive test result has or will have the phenotype for which the marker is believed to be a predictor. Confirmed clinical validity does not necessarily mean that a biomarker can immediately be used in patient care. This is achieved in the next clinical utility step. Only biomarker candidates with confirmed analytical and clinical validity are submitted to the formal assessment of clinical utility. In this step, large, independent, and well-designed studies evaluate the biomarker’s expected utility using formal statistical tools. It is this stage that is usually underappreciated in discussions of the value of a marker as a biomarker. Only after passing this step, a marker attains a biomarker status with clinical utility meaning that it provides information that is useful in the clinic in making decisions about disease susceptibility, disease classification, prognostic stratification, or response to a given treatment. Once evidence of utility in real clinical settings is generated, the regulatory process begins for introduction of the biomarker for use in routine clinical care (Fig. 2).
Evaluation of a Biomarker for Clinical Utility
A biomarker is a classifier that classifies a group of people into separate groups like susceptible and non-susceptible individuals, one subgroup of a disease and another, or a subgroup with good prognosis and another without. The clinical utility of a biomarker is assessed in a different manner from the way a case–control study evaluates an association. Here the focus is on whether by using a biomarker subjects can be reclassified in a class different from where they would be classified using existing evidence. An ideal biomarker, say, for disease susceptibility, with its high specificity and sensitivity (positive in every predisposed subject and negative in every subject that will remain disease-free, thus, yielding a high likelihood ratio), is well calibrated (the predicted risk for developing the disease corresponds to real risk and not over- or underestimated) and therefore would classify every subject as predisposed or not. Once this ideal stage is reached, the positive and negative predictive values for the marker will also be ideal demonstrating the benefits and risks from both positive and negative results. Such an imaginary biomarker would have a value of 100 % for each of sensitivity, specificity, and area under “ROC” curve (AUC). This scenario is only true for some monogenic Mendelian disorders where the genetic effect is not modified by the environment.
In the ultimate stage of biomarker development preceding the regulatory approval, statistical evaluation is most rigorous. Logistic regression and its output of effect size, the OR, are not good indicators of the clinical utility of the biomarker candidate. The OR is not an estimate of individual-level risk nor a diagnostic test or classifier; and the OR should not be used in a predictive study as the effect size [41]. A high OR (or relative risk) associated with a marker even after adjusting for established risk factors does not necessarily translate into better risk prediction. The statistical considerations differ between etiologic risk studies and studies on biomarkers that are going to be used to classify subjects. Studies that use the OR as the effect size use multiple markers with small effect sizes simultaneously to increase the effect size. This approach certainly increases the effect size, but the multimarker set is still not a good classifier. In a simulation study of 40 independent genetic risk markers each yielding an odds ratio of 1.2–2.0 and a sample size of one million, the best results in discriminative accuracy quantified as the AUC were obtained when the risk genotypes were all common (≥30 %) and odds ratios were closer to 2.0. Even with this unrealistic scenario, the AUC was 0.93 [42]. This example shows that even simultaneous use of most common and strong risk markers may still not have a large discriminatory value between disease and healthy states.
The ROC curve analysis is a commonly used measure of performance of a predictive test. An ideal marker has an AUC of 1 representing perfect discrimination between the diseased and nondiseased subjects; in other words, all subjects are correctly classified by the test. The baseline value for an AUC is 0.5 which represents no discrimination at all; in other words subjects are classified no more correctly than can be attributed to by chance. AUC plots the sensitivity of the marker against (1 – specificity) for all possible cutoff values. In the case of a binary marker, this is just a single point. The c-index is numerically equivalent to the AUC. A serious issue with the AUC is that it does not measure the ability of a new marker to add value to a preexisting prediction model. Thus, a new marker may have a good performance, but whether adding the new marker to existing markers will improve the performance needs to be known. This is usually done by generating two ROC curves, one with and one without the new marker using information on existing markers, and observing whether there is a difference between them. The difference can be formally assessed by the c-statistics if the change in AUC appears to be substantial enough.
However, useful ROC curves may be for classification; evaluation of predictive models cannot rely solely on the ROC curve, but should assess discrimination and calibration using new metrics (Table 1). Jakobsdottir et al. looked at this formally [16]. They compared the utility of genetic markers of a number of complex disorders identified by GWAS using ROC curve analysis against conventional risk markers already known and concluded that small P values, high ORs, and AUCs do not guarantee good prediction of actual risk. The AUC should be considered as a first step in evaluating a model or in comparing two models against each other. The AUC value is, however, insufficient on its own to show that a model would improve decision-making. One criticism aimed at the AUC analysis in clinical utility determination is that it gives equal weight to specificity and sensitivity, hence to false positives and false negatives, which may not be the case in a real clinical situation [43]. Published estimates of disease prevalence and heritability or sibling recurrence risk for 17 complex genetic diseases have been used to calculate the proportion of genetic variance that a test must explain to achieve an AUC = 0.75, which is a modest value. For 17 diseases, the proportion of genetic variance that have to be explained by genetic markers for the predictive model to attain an AUC value of 0.75 varied from 0.10 to 0.74. In other words, depending on disease prevalence and heritability, genetic markers can explain as little as 0.10 or as high as 0.74 of the heritability to yield an AUC value of 0.75, the threshold regarded as making a diagnostic classifier clinically useful when applied to a sample considered to be at increased risk [42]. On the other hand, a threshold AUC value is 0.99 for a predictive test to be a classifier when applied to the general population. Given the prevalence and heritability of RA, the maximum value for genetic markers in RA for prediction of disease susceptibility can get close (0.98) but not quite reach this threshold [48].
Table 1
Comparison of models for risk prediction
Association | Logistic regression |
|---|---|
Global model fit | Likelihood ratio test; Bayes information criterion |
Discrimination | ROC; concordance (or c)-statistics |
Calibration | Hosmer–Lemeshow statistic for goodness of fit |
Risk reclassification | Integrated discrimination improvement (IDI); net reclassification improvement (NRI); category-free NRI (cfNRI); decision curve analysis |
For clinical utility assessment, additional statistical methods including discrimination metrics (c-statistics), measures of calibration (addressing how close the predicted risks are to the actual observed risks), and reclassification (addressing whether the model including the novel biomarker changes a person’s risk sufficiently to move them to a different risk category) are needed (Table 1) [44–46, 49, 50]. Calibration is essential for good decision-making. A model is well calibrated when the predicted risk is equal to the observed risk. Calibration takes into account the average risk in a population. Although essential in biomarker development, calibration is not sufficient for clinical utility. What is most crucial for determination of clinical utility in biomarker development is reclassification, which aims to do at the individual level what AUC analysis does at the group level. In reclassification analysis, each individual’s data is considered to see whether the new marker changes their risk classification. This is achieved by reclassification tables that show changes in individual risk classifications with the addition of the new marker to the prediction model. It has been pointed out that a marker which has only modest or no effect by AUC analysis may still improve risk classification at the individual level [44, 45].
Past Mistakes in Biomarker Development
It is important to learn from past mistakes for current efforts in biomarker development to be more productive. It is well known that most claims of classification performance are overly optimistic, lacking verification by replication with questionable generalizability. Ioannidis reviewed the most common causes of biomarker failures [51]. He has pointed out that despite the introduction of so many biomarkers into clinical practice, the health impacts have not been favorable in general. Four types of failures have been recognized as summarized in Table 2. The most dramatic of these failures concerns the PSA. Initially introduced in the 1980s for monitoring treatment response, PSA testing has found a place as a biomarker for screening and early diagnosis of prostate cancer [52]. The AUC for PSA in ROC curve analysis is 0.68 for cancer versus no cancer. Given that a test that performs no better than chance yields an AUC of 0.5, this value does not suggest a high clinical utility in classifying subjects as having prostate cancer or not. PSA is believed to have done more harm and good, by drastically increasing overdiagnosis and overtreatment of prostate cancer [53]. The problem with PSA was that it was not subject to a robust assessment before being used for screening and actual clinical use disappointed. A search for biomarkers is still ongoing. The PSA example illustrates the importance of following the steps shown in Fig. 2 before introducing a biomarker to clinical use.
Table 2
Types of biomarker failures
Type A | Type B | Type C | Type D | |
|---|---|---|---|---|
Problem | A biomarker makes it to the clinics, but does not fulfill the promise | A biomarker is reported to have strong features, but cannot be validated by following studies | A biomarker is found in one study, but clinical optimization is lacking | A biomarker is promoted despite lack of promising evidence |
Example | Prostate-specific antigen (PSA) | Proteomic markers of ovarian cancer | Gene expression signatures in cancer | Direct-to-consumer genetic risk determination |
Solution | Satisfactory assessment of clinical utility before introducing it into practice | Verification of analytical validity before proceeding with the development | Use of robust statistical methods at the development phase and follow-up toward clinical implementation | Better assessment of incremental benefit of using these markers over existing risk markers |
An ideal biomarker for clinical use should have three major characteristics: (1) It should be safe and easy to measure preferably noninvasively and with good reproducibility; (2) it should have a high sensitivity, high specificity, and high positive and negative predictive values (PPV and NPV, respectively) for its intended outcome; and (3) it should improve decision-making abilities in line with clinicopathological parameters. Any attempt to introduce a single marker to routine care as a biomarker will probably suffer one of the types of failure listed in Table 2. If a single biomarker cannot fulfill all the expectations, which is usually the case, a panel of multiple biomarkers if performing better than any single biomarker may be used. Indeed, most common biomarkers used in RA to assess disease activity (multi-biomarker disease activity or MBDA) are of this type.
Validity of Existing Biomarkers for Rheumatoid Arthritis
In RA, associations of markers, especially those that are genetic, have been widely reported as valid for a number of outcomes, but the development of biomarkers based on those findings has been either unsuccessful or slow. As discussed above, a high OR is no indication of success for a biomarker, and the strongest HLA region association in RA has an OR of 5–10. Even the strongest HLA association in any disease with an OR of around 100 as in ankylosing spondylitis may not turn out to be a good biomarker. This highlights the difficulty with converting even the strongest risk markers to biomarkers.
As discussed below, there are a number of biomarkers used for RA diagnosis, disease activity, assessment, or prognosis. A survey analyzed the validity of biomarkers as reported in 170 articles [40]. Most common biomarkers were gene expression profiles. Flaws were identified in most reports. Less than half of the studies incorporated study-design features important for valid clinical associations: age and sex-matched groups and controlling for medications used. These issues concerned mainly the discovery stage studies. Even at that stage, which forms the foundation of a long process, no more than half of the studies were satisfactory by simple epidemiologic criteria. This is not a promising start to the process of biomarker development if biomarkers with genuine clinical utility are the aim.
To avoid future mistakes in biomarker development and to aid with valid ones, an independent initiative called OMERACT (Outcome Measures in Rheumatology) consisting of international health professionals interested in outcome measures in rheumatology was formed in 1992. OMERACT has played a critical role in the development and validation of clinical and radiographic outcome measures in RA and other rheumatic diseases. A special interest group developed validation criteria for soluble biomarkers of structural joint damage [36]. These criteria have been further developed and put to test for existing biomarkers [54–57]. Neither a baseline C-reactive protein (CRP) test nor later tests on five more soluble biomarkers receptor activator of nuclear factor kappa-B ligand (RANKL; TNFSF11), osteoprotegerin (OPG), matrix metalloprotease (MMP-3), and urine C-telopeptide of types I and II collagen (U-CTX-I and U CTX-II) produced strong evidence that these biomarkers could substitute for radiographic endpoints in RA. The OMERACT validation criteria are based on three domains: truth (is the measure truthful, does it measure what it intends to measure? Is the result unbiased and relevant?), discrimination (does the measure discriminate between situations that are of interest?), and feasibility (can the measure be applied easily, given constraints of time, money, and interpretability?) This initiative together with other published guidelines for various aspects of biomarker development studies are expected to result in the development of reliable biomarkers with enhanced validity and clinical utility.
Biomarkers in RA
Earliest Biomarkers in RA
Like many other complex disorders, RA is a heterogeneous disease. Traditionally, RA has been classified as rheumatoid factor (RF) positive and RF negative, but more recently, antibodies to citrullinated protein antigen (ACPA), also referred to as anti-CCP (Anti-cyclic citrullinated protein), are used in the classification of RA. Of the two, ACPA is more specific to RA as RF is more likely than ACPA to be positive in other rheumatic disorders [58]. These autoantibodies were therefore the earliest biomarkers for RA diagnosis and classification (Table 3). It is very important to take this heterogeneity into account in any study, but especially in studies of primary susceptibility. Thus, biomarkers should be developed for these subtypes separately. ACPA has different fine specificities, but they do not seem to provide additional information regarding the clinical phenotype at present. Isotype usage in ACPA response increases to include more diverse antibodies (IgM, IgA, all IgG subclasses, and IgE) together with the titer of ACPA before the development of full-blown disease [59]. In contrast, isotype usage does not change once RA settles as the full-blown disease.
Table 3
Disease classification by ACPA antibody status
Characteristic | ACPA positive | ACPA negative |
|---|---|---|
Heritability | ~60 % | ~60 % |
Disease course | Severe | Milder |
Drug-free remission probability | Lower | Greater |
HLA (shared epitope) association | Yes (HLA–DRB1*01; *04) | No |
Other HLA associations | HLA–DRB1*15 | HLA–DRB1*03; *13 |
PTPN22 association | Yes | No |
Other genetic associations | CTLA4, STAT4, PADI4, CTLA4, TNFAIP3–OLIG3, TRAF1/C5, FCGR, IL2RA, IL2RB, CD40, CTL21, CCR6, and others | IRF5, STAT4 |
Smoking association | Yes and only in HLA shared epitope-positive subjects | No |
More recently, another antibody specificity has been identified in RA. Anti-carbamylated protein (anti-CarP) antibodies are formed against homocitrullinated proteins with little or no cross-reactivity to ACPA [59]. Similar to ACPA positivity, anti-CarP antibodies have been proposed to predict the development of RA in patients with undifferentiated arthritis or arthralgia and predict joint damage [10, 59–61]. Anti-CarP antibodies may be detected in both ACPA-positive and ACPA-negative RA patients, and the unfavorable effect on the clinical course is more prominent in ACPA-negative patients [60]. More specific and sophisticated versions of the earliest autoantibodies are now available especially for early diagnosis of RA in patients who present with arthritis and will be discussed below.
Biomarkers for Disease Susceptibility
Genetic Markers for Disease Susceptibility
To examine genetic markers for disease susceptibility, it should be first established that the disease has genetic background. In the case of RA, this is well established. Both early candidate gene studies [62] and a major GWAS [63] have provided strong evidence for genetic susceptibility to RA (reviewed in [64]) (Table 4). Stronger evidence comes from classical twin studies which have estimated that heritability of RA exceeds 50 % [19–21]. The concordance rate for RA among monozygotic twins is higher in all studies (12–15 %) than among dizygotic twins (4 %) [19, 20] although heritability estimates based on familial resemblance are a little lower [22]. In twin studies, heritability estimates between ACPA-positive and ACPA-negative RA do not differ much: 68 % vs. 66 % [20]. Genetic contribution in similar magnitude has also been documented for the progression of joint damage in RA [79]. While it is clear that there is sizeable genetic contribution, environmental contribution is probably equally large. In another study of 13 monozygotic twin pairs discordant for RA and smoking, in 12 of 13 pairs, the smoking twin member was also the proband [80]. This example shows the importance of considering genetic and environmental factors in any study investigating susceptibility to RA. Besides smoking, alcohol consumption is an important risk modifier for RA. On the other hand when it comes to disease progression, countries where smoking is more prevalent, i.e., Turkey, severity of disease seems less.
Table 4
Genetic modifiers of RA susceptibility identified in GWAS and meta-analysis of multiple GWAS currently listed in GWAS catalog
Chromosome region | Chromosome position | Gene | SNP | P value | Odds ratio (95 % CI) | Population | Features | Reference |
|---|---|---|---|---|---|---|---|---|
6p21.1 | 44232920 | NFKBIE | rs2233434 | 1E–15 | 1.20 (1.15–1.26) | Japanese | Double hita | [65] |
10q21.2 | 63958112 | RTKN2 | rs3125734 | 5E–9 | 1.20 (1.13–1.27) | Japanese | [65] | |
2p15 | 62452661 | B3GNT2 | rs11900673 | 1E–8 | 1.11 (1.07–1.15) | Japanese | [66] | |
4q21.21 | 79513215 | ANXA3 | rs2867461 | 1E–12 | 1.13 (1.09–1.17) | Japanese | [66] | |
5q31.1 | 131430118 | CSF2 | rs657075 | 3E–10 | 1.12 (1.08–1.15) | Japanese | [66] | |
6p23 | 14096658 | CD83 | rs12529514 | 2E–8 | 1.14 (1.09–1.19) | Japanese | [66] | |
6p21.1 | 44232920 | NFKBIE | rs2233434 | 6E–19 | 1.19 (1.15–1.24) | Japanese | Double hita | [66] |
10q21.2 | 63785089 | ARID5B | rs10821944 | 6E–18 | 1.16 (1.12–1.20) | Japanese | [66] | |
11q13.4 | 72373496 | PDE2A, ARAP1 | rs3781913 | 6E–10 | 1.12 (1.08–1.16) | Japanese | [66] | |
14q32.33 | 105391005 | PLD4 | rs2841277 | 2E–14 | 1.15 (1.11–1.19) | Japanese | [66] | |
18p11.21 | 12797694 | PTPN2 | rs2847297 | 2E–8 | 1.10 (1.07–1.14) | Japanese | [66] | |
11q24.3 | 128492739 | ETS1, FLI1 | rs4937362 | 8E–7 | 1.09 (1.06–1.13) | Japanese | [66] | |
14q22.2 | 55348118 | GCH1 | rs3783637 | 2E–6 | 1.10 (1.06–1.14) | Japanese | [66] | |
14q23.1 | 61908332 | PRKCH | rs1957895 | 4E–7 | 1.09 (1.05–1.13) | Japanese | [66] | |
15q26.1 | 90893668 | ZNF774 | rs6496667 | 1E–6 | 1.09 (1.05–1.13) | Japanese | [66] | |
16p12.2 | 23888840 | PRKCB1 | rs7404928 | 4E–6 | 1.08 (1.05–1.12) | Japanese | [66] | |
16q24.1 | 86018633 | IRF8 | rs2280381 | 2E–6 | 1.12 (1.07–1.17) | Japanese | [66] | |
6p21.33 | 31622606 | APOM | rs805297 | 3E–10 | 1.56 (1.36–1.80) | Korean | HLA region | [67] |
6p21.32 | 32429643 | HLA-DRA | rs9268853 | 5E–109 | 2.40 (2.20–2.60) | European | HLA region | [68] |
6p21.32 | 32602269 | HLA-DQA1 | rs9272219 | 1E–45 | 1.92 (1.75–2.08) | European | HLA region | [68] |
6p21.33 | 31379931 | MICA | rs1063635 | 1E–17 | 1.35 (1.27–1.45) | European | HLA region | [68] |
6p22.1 | 29789171 | HLA-G | rs1610677 | 4E–15 | 1.32 (1.79–1.41) | European | HLA region | [68] |
22q12.3 | 37551607 | IL2RB | rs743777 | 2E–6 | 1.19 (1.10–1.30) | European | [68] | |
21q22.3 | 45709153 | AIRE, PFKL | rs2075876 | 4E–9 | 1.18 (1.11–1.24) | Japanese | [69] | |
6p21.32 | 32218989 | NOTCH4 | rs9296015 | 2E–38 | NR | Japanese | HLA region | [69] |
1p36.13 | 17674537 | PADI4 | rs2240335 | 2E–8 | NR | Japanese | Only observed in Asians; double hita | [69] |
8p23.1 | 11359638 | BLK | rs1600249 | 5E–6 | 1.30 (1.16–1.45) | Korean | [70] | |
12q21.1 | 72724034 | TRHDE | rs12831974 | 6E–6 | 1.27 (1.14–1.40) | Korean | [70] | |
3p14.3 | 56966246 | ARHGEF3 | rs2062583 | 2E–6 | 1.59 (1.30–1.92) | Korean | [70] | |
6p21.32 | 32680928 | HLA-DRB1 | rs7765379 | 5E–23 | 2.51 (NR) | Korean | HLA region | [70] |
1p36.13 | 17674537 | PADI4 | rs2240335 | 2E–8 | 1.50 (NR) | Korean | Only observed in Asians; double hita | [70] |
6q27 | 167532793 | CCR6 | rs3093024 | 8E–19 | 1.19 (1.15–1.24) | Japanese | [71] | |
6p21.32 | 32671103 | HLA-DRB1 | rs13192471 | 2E–58 | 1.97 (1.82–2.14) | Japanese | HLA region | [71] |
2q32.3 | 191964633 | STAT4 | rs7574865 | 2E–6 | 1.17 (1.10–1.25) | Japanese | ACPA-positive and ACPA-negative disease; double hita | [71] |
6q23.3 | 138196066 | OLIG3,TNFAIP3 | rs2230926 | 2E–6 | 1.31 (1.17–1.46) | Japanese | ACPA-positive and ACPA-negative disease | [71] |
2p14 | 65595586 | SPRED2 | rs934734 | 5E–10 | 1.13 (NR) | European | [72] | |
5q11.2 | 55438580 | ANKRD55,IL6ST | rs6859219 | 1E–11 | 1.28 (NR) | European | [72] | |
5q21.1 | 102596720 | C5orf30 | rs26232 | 4E–8 | 1.14 (NR) | European | ACPA-positive and ACPA-negative disease | [72] |
3p14.3 | 58556841 | PXK | rs13315591 | 5E–8 | 1.29 (NR) | European | [72] | |
4p15.2 | 26108197 | RBPJ | rs874040 | 1E–16 | 1.14 (NR) | European | [72] | |
6q27 | 167534290 | CCR6 | rs3093023 | 2E–11 | 1.13 (NR) | European | [72] | |
7q32.1 | 128594183 | IRF5 | rs10488631 | 4E–11 | 1.19 (NR) | European | Stronger in ACPA-negative disease | [72] |
2q11.2 | 100806940 | AFF3 | rs11676922 | 1E–14 | 1.12 (NR) | European | [72] | |
9p13.3 | 34743681 | CCL21 | rs951005 | 4E–10 | 1.19 (NR) | European | [72] | |
10p15.1 | 6098949 | IL2RA | rs706778 | 1E–11 | 1.14 (NR) | European | [72] | |
1q24.2 | 167408670 | CD247 | rs840016 | 2E–6 | 1.11 (NR) | European | [72] | |
4q27 | 123218313 | IL2,IL21 | rs13119723 | 7E–7 | 1.12 (NR) | European | [72] | |
12q24.12 | 111884608 | SH2B3 | rs3184504 | 6E–6 | 1.08 (NR) | European | [72] | |
14q24.3 | 75960536 | BATF | rs7155603 | 1E–7 | 1.16 (NR) | European | [72] | |
17q12 | 38040763 | IKZF3 | rs2872507 | 9E–7 | 1.10 (NR) | European | [72] | |
21q22.3 | 43836186 | UBASH3A | rs11203203 | 4E–6 | 1.11 (NR) | European | [72] | |
1p36.32 | 2553624 | TNFRSF14 | rs3890745 | 4E–6 | 1.12 (1.06–1.18) | European | [72] | |
1p13.2 | 114377568 | PTPN22 | rs2476601 | 9E–74 | 1.94 (1.81–2.08) | European | Strongest non-HLA risk marker; double hita | [72] |
2p16.1 | 61136129 | REL | rs13031237 | 8E–7 | 1.13 (1.07–1.18) | European | [72] | |
2q11.2 | 100835734 | AFF3 | rs10865035 | 2E–6 | 1.12 (1.07–1.17) | European | [72] | |
2q32.3 | 191964633 | STAT4 | rs7574865 | 3E–7 | 1.16 (1.10–1.23) | European | ACPA-positive and ACPA-negative disease; double hita | [72] |
2q33.2 | 204738919 | CTLA4 | rs3087243 | 1E–8 | 1.15 (1.10–1.20) | European | [72] | |
6q23.3 | 138006504 | TNFAIP3 | rs6920220 | 9E–13 | 1.22 (1.16–1.29) | European | [72] | |
9q33.2 | 123690239 | TRAF1,C5 | rs3761847 | 2E–7 | 1.13 (1.08–1.18) | European | Double hita | [72] |
10p15.1 | 6393260 | PRKCQ | rs4750316 | 2E–6 | 1.15 (1.09–1.22) | European | Double hita | [72] |
20q13.12 | 44747947 | CD40 | rs4810485 | 3E–9 | 1.18 (1.11–1.25) | European | [72] | |
1p34.3 | 38624129 | POU3F1 | rs12131057 | 4E–7 | 1.16 (NR) | European | [72] | |
15q23 | 69995344 | KIF3 | rs17374222 | 2E–6 | 1.13 (NR) | European | [72] | |
6p21.32 | 32282854 | HLA-DRB1 | rs6910071 | 1E–299 | 2.88 (2.73–3.03) | European | HLA region | [72] |
2p16.1 | 61164331 | REL | rs13017599 | 2E–12 | 1.21 (1.15–1.28) | European | [73] | |
2q33.2 | 204693876 | CTLA4 | rs231735 | 6E–9 | 1.17 (1.11–1.23) | European | [73] | |
8p23.1 | 11343973 | BLK | rs2736340 | 6E–9 | 1.19 (1.13–1.27) | European | [73] | |
1p13.2 | 114377568 | PTPN22 | rs2476601 | 2E–21 | NR | European | Strongest non-HLA risk marker; double hita | [73] |
9q33.2 | 123652898 | TRAF1, C5 | rs881375 | 4E–8 | NR | European | [73] | |
1p36.32 | 2553624 | MMEL1,TNFRSF14 | rs3890745 | 1E–7 | 1.12 (NR) | European | [74] | |
7q21.2 | 92246744 | CDK6 | rs42041 | 4E–6 | 1.11 (NR) | European | [74] | |
9p13.3 | 34710260 | CCL21 | rs2812378 | 3E–8 | 1.12 (NR) | European | [74] | |
12q13.3 | 57968715 | KIF5A,PIP4K2C | rs1678542 | 9E–8 | 1.12 (NR) | European | [74] | |
20q13.12 | 44747947 | CD40 | rs4810485 | 8E–9 | 1.15 (NR) | European | [74] | |
10p15.1 | 6393260 | PRKCQ | rs4750316 | 4E–6 | 1.14 (NR) | European | Double hita | [74] |
1p13.2 | 114303808 | PTPN22 | rs6679677 | 6E–42 | 1.79 (1.65–1.94) | European | Double hita | [74] |
6p21.32 | 32663999 | HLA-DRB1 | rs6457620 | 4E–186 | 2.55 (2.40–2.71) | European | HLA region | [74] |
6q23.3 | 138006504 | OLIG3, TNFIP3 | rs6920220 | 2E–9 | 1.24 (1.16–1.32) | European | [74] | |
18q23 | 76409597 | SALL3 | rs2002842 | 6E–6 | 1.61 (NR) | Spanish | [75] | |
6p21.32 | 32663851 | HLA-DQA1, HLA-DQA2 | rs6457617 | 1E–9 | NR | Spanish | HLA region | [75] |
6q23.3 | 138002637 | TNFAIP3, OLIG3 | rs10499194 | 1E–9 | 1.33 (1.15–1.52) | European | [76] | |
6q23.3 | 138006504 | TNFAIP3, OLIG3 | rs6920220 | 1E–7 | 1.22 (NR) | European | [76] | |
9q33.2 | 123690239 | TRAF1-C5 | rs3761847 | 4E–14 | 1.32 (1.23–1.42) | European | Double hita | [77] |
1p13.2 | 114377568 | PTPN22 | rs2476601 | 2E–11 | 1.72 (NR) | European | Strongest non-HLA risk marker; double hita | [77] |
6p21.32 | 32577380 | HLA-DRB1 | rs660895 | 1E–108 | 3.62 (NR) | European | HLA region | [77] |
7q32.3 | 131370039 | Intergenic | rs11761231 | 4E–7 | 1.32 (NR) (women) | European | [78] | |
22q12.3 | 37551607 | NR | rs743777 | 1E–6 | 1.09 (0.97–1.24) | European | [78] | |
21q22.2 | 42511918 | NR | rs2837960 | 2E–6 | 1.05 (0.93–1.20) | European | [78] | |
4p15.2 | 25417244 | NR | rs3816587 | 9E–6 | 1.09 (0.96–1.25) | European | [78] | |
6p21.32 | 32574171 | HLA-DRB1 | rs615672 | 8E–27 | NR | European | HLA region | [78] |
1p13.2 | 114303808 | PTPN22 | rs6679677 | 6E–25 | 1.98 (1.72–2.27) | European | Double hita | [78] |
6p21.32 | 32663851 | HLA-DQB1 | rs6457617 | 5E–75 | 2.36 (1.97–2.84) | European | HLA region | [78] |
Alcohol reduces the risk for RA as well as joint damage measured by X-ray [81].
As ACPA-positive disease makes up around 70 % of all RA cases, most genetic association studies and almost all major GWAS have been conducted in ACPA-positive cases. The largest study ever conducted in ACPA-negative cases only examined known risk markers for ACPA-positive disease at the time of the study [82]. Thus, there is still need to examine risk markers exclusive to ACPA-negative disease. In fact, this approach is likely to be fruitful since heritability estimates for ACPA-positive and ACPA-negative RA are similar at least in twin studies, but the contribution of HLA complex to these estimates is much higher in ACPA-positive RA [20]. Thus, ACPA-negative RA is expected to have non-HLA markers stronger than those observed in ACPA-positive RA. The recently identified subtype characterized by anti-CarP antibodies has yet to be examined for genetic associations.
Although there have been a large number of candidate gene studies and have shown associations with RA susceptibility, RA is one of those diseases that have been most extensively studied by GWAS. Genome-wide association studies conducted in large discovery and replication samples have identified more than 45 confirmed associations (Table 4). After the first-generation GWAS, cumulative results have been subjected to meta-analyses [66, 72, 83, 84] and finally the Immunochip custom SNP array analysis [85]. Overall GWAS results indicate that heritability of RA is more than 50 %, of which HLA explains 36 % [21, 85]. This estimate of the contribution of HLA to RA heritability is considerably higher than a previous estimate based on a twin study in which the presence of the HLA shared alleles explained 18 % of the genetic variance of ACPA-positive RA but only 2.4 % of the genetic variance of ACPA-negative RA [20]. Anti-CCP development has also been examined for its genetic associations [86]. The strongest associations map to the HLA region to the class III and class II region border. The statistically most significant result was achieved by rs1980493 (P = 6 × 10−5). This association was still strong after adjustment for the presence of the shared epitope. This SNP is in an intergenic region between BTNL2 and HLA–DRA. Bioinformatic analysis shows its involvement in transcriptional and splicing regulation (functionality score = 0.50; range = 0–1).
The original HLA association was with HLA-Dw4 corresponding to HLA-DR4 [62]. It was refined to be the shared epitope (QRRAA, RRRAA, and QKRAA) encoded by the amino acids in positions 70–74 of HLA-DRβ1 molecule [87]. Further exploration of the “shared epitope” revealed that the association of the RAA sequence occupying positions 72–74 is modulated by the amino acids at positions 71 and 72. At position 71, K confers the highest risk, R an intermediate risk, A and E a lower risk; and at position 70, Q or R confers a higher risk than D [88]. The shared epitope predisposes an individual to ACPA production and in interaction with smoking [89, 90]. The association of HLA-DR shared epitope with the disease itself, and its clinical development is secondary to its association to anti-CCP [89]. In ACPA-negative cases, HLA shared epitope shows no association, but instead HLA–DRB1*03 is a risk factor [91]. Besides the shared epitope association in ACPA positive with risk of RA mediated by ACPA production, HLA–DRB1*1301 is also associated with RA but with protection [92].
In the most recent study of RA associations with individual amino acids positions in 5,014 ACPA-positive cases and almost 15,000 controls, three positions (11, 71, and 74) in HLA-DRβ1 and two in other HLA proteins (position 9 in HLA-B and position 9 in HLA-DPβ1) appeared to explain the risk conferred by the HLA complex [93]. All these positions are located in the peptide-binding grooves and suggest that HLA associations are causally related to peptide presentation function of HLA molecules although shared epitope is implicated in signal transduction too [94]. The most significant association within the HLA region is with the imputed SNP rs17878703 (allele A), a quadrallelic SNP in the second nucleotide of DRB1 codon 11 (OR = 3.7, P <10−526) [93]. As this SNP is in one of the most polymorphic regions in the genome and quadrallelic (i.e., all four nucleotides are alleles of this SNP), it is not included in current genotyping platforms for GWAS due to technical difficulties and has to be imputed.
In the largest meta-analysis of more than 2.5 million SNPs in 5,539 RA cases and 20,169 controls of European descent, the top five ranking candidate causal SNP associations were all from the HLA complex (rs1063478, rs375256, rs365066, rs2581, and rs1059510) [84]. These results were subjected to pathway analysis to learn more about disease biology. HLA region associations contributed to the most strongly associated pathway. The HLA–DMA SNP rs1063478 is a missense variant (V166I) and alters the role of HLA-DM protein in antigen processing and presentation. Together with the peptide-binding groove polymorphisms, overall results in the HLA region implicate the antigen processing and presentation as the major biological pathway in the pathogenesis of RA. The HLA–DRB1 association is not only the strongest for RA susceptibility but is also associated with systemic forms of RA [95, 96] and with radiologic damage [97].
Among the non-HLA region associations, that of protein tyrosine phosphatase non-receptor 22 gene (PTPN22) is outstanding [98]. The SNP rs2476601 (R620W) alters the role of the PTPN22 protein in the context of immune response-activation cell surface receptor signaling pathway. The PTPN22 association is exclusive to European populations as the risk allele is either absent or very rare in Asians and Africans. Interestingly, these populations respond to the same treatments for RA, and phenotypically the disease is indistinguishable. Other noteworthy associations are with CD40, STAT4, PRM1, PADI4, TRAF1/C5, and TNFAIP3 variants (Table 4). The tumor necrosis factor-alpha-induced protein 3 (TNFAIP3) SNP rs2230926 (F127C) alters the role of TNFAIP3 in the context of the CD40L signaling pathway [84, 99]. The PADI4 association is strongest in Asian populations [100]. PADI4 encodes the type 4 peptidylarginine deiminase enzyme, which posttranslationally converts peptidylarginine to citrulline, generating citrullinated proteins. Its association with RA risk is, therefore, biologically plausible. Of the genetic associations, IRF5 association is exclusive to anti-CCP-negative subset of RA [101]. It has been, however, proven difficult to unravel genetic associations exclusive to ACPA-negative subset either by a case–control or a case-only design [82, 102]. The case-only design which compared ACPA-positive with ACPA-negative cases noted that the largest difference between the two subtypes lies within the HLA complex [102].
Related posts:
 Medicine in Rheumatology: How Does It Differ from Other Diseases?
Measures in Rheumatoid Arthritis
Classification/Diagnosis Criteria
Randomized Controlled Trial: Methodological Perspectives
Reviews and Meta-analyses in Rheumatology
of Traditional Randomized Controlled Clinical Trials in Rheumatology
Medicine in Rheumatology: How Does It Differ from Other Diseases?
Measures in Rheumatoid Arthritis
Classification/Diagnosis Criteria
Randomized Controlled Trial: Methodological Perspectives
Reviews and Meta-analyses in Rheumatology
of Traditional Randomized Controlled Clinical Trials in Rheumatology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


