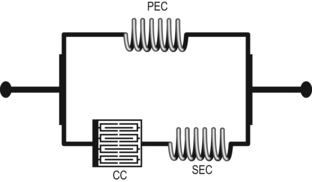
4 In 1861, William Little wrote the first medical descriptions of a disorder, named Little disease, that caused stiff, spastic muscles in the legs of affected young children (Bax et al., 2005; Jones et al., 2007; Shimony et al., 2008). Little disease is now known as the diplegia form of cerebral palsy (CP) (Pincus, 2000). A century later, Bax (1964) defined CP as ‘a disorder of posture and movement due to a defect or lesion of the immature brain’. More recently Bax et al. (2005), through the American Academy for Cerebral Palsy and Developmental Medicine, proposed a new definition: It is currently this definition that is most often used. CP is not a disease, per se, but rather, a term that describes a heterogeneous condition resulting in chronic motor impairment and other impairments. Although the brain lesion may be non-progressive, variable or progressive symptoms also occur due to the effects of growth and development (Eames et al., 1997; Klingbeil et al., 2004) and of adaptive changes occurring in the neuromusculoskeletal system (Carr and Shepherd, 1998; Gajdosik and Cicirello, 2001). The movement disorders resulting from the lesion and from adaptive changes occurring as the infant grows and develops become more evident and more severe over time (Hanna et al., 2009). This progression seems to occur in particular as a result of adaptive changes occurring in muscle (Friden and Lieber, 2003; Malaiya et al., 2007; Mohagheghi et al., 2007). Children with CP are already at a disadvantage, their levels of physical functioning having a lower starting point due to slowed development of muscle, bone and cardiorespiratory system following the insult to the brain (Damiano, 2006). These children are usually less physically active than typically developing children, and their physical activity levels tend to decrease with increasing age (Maher et al., 2007). Worldwide, CP is one of the most common causes of chronic disability in children. In Europe, the prevalence has risen from about 1.5 per 1000 live births in the 1960s to about 2.5 in the 1990s (Odding et al., 2006). However the overall prevalence of CP remains stable (Baxter, 2009), occurring in 2–2.5 individuals per 1000 live births in developed countries (Blair and Stanley, 1997; Hirtz et al., 2007; Odding et al., 2006; Sigurdardottir et al., 2009; Stanley et al., 2000). The worldwide cumulative incidence rate at 5–7 years was 2.7 cases of CP for 1000 birth cohorts, with approximately 36% of all CP occurring in infants weighing less than 2500 g at birth (Rosen and Dickinson, 1992). Improvement in the survival rate of infants with a low birth weight is accompanied by an increase in the rate of neurological impairment or disability among the survivors (Escobar et al., 1991; Kitchen et al., 1992). The prevalence of CP is higher among both the low birth weight children (Odding et al., 2006) and preterm-born children (Himpens et al., 2008). Some form of motor impairment is present in all children with CP, with the majority (72–91%) reported to display spasticity (Odding et al., 2006). One study has found that 73% of children and adolescents with a spastic hemiplegic presentation also have soft tissue contractures (Odding et al., 2006). Multiple risk factors can be identified in over 70% of cases (Bialik and Givon, 2009; Jones et al., 2007). These risk factors cause injury to the developing brain and can occur in the prenatal (38%) or peri/neonatal (35%) periods (uncertain in 27%) in children born at term. In preterm children, brain injury is less frequent prenatally (17%) and more common in the perinatal period (49%) or occurs at an uncertain time (33%) (Himmelmann et al., 2005). Children with CP may have one or more risk factors, with the definitive cause difficult to determine (Jones et al., 2007). Prenatal risk factors are associated with infection, maternal drug or alcohol abuse, genetic causes, maternal epilepsy, mental retardation, hyperthyroidism, severe toxaemia, third trimester bleeding and low birth weight (less than 1500 g) (Pakula et al., 2009). Perinatal risk factors are associated with hypoxia or birth trauma such as brain haemorrhage during delivery and placental complications. Postnatal causes include head trauma, meningitis, encephalitis and brain infarcts (Bialik and Givon, 2009; Jones et al., 2007). The evidence on lesion site from recent neuroimaging studies has consistently shown abnormalities in 70–90% of affected children, facilitating clinical classification into groups with early brain malformations, periventricular white matter injury, neonatal encephalopathies and focal ischaemic ⁄ haemorrhagic lesions (Hoon, 2005; Robinson et al., 2009; Wu et al., 2006). Using neuroimaging studies can assist clinicians in the early identification of injury before the establishment of marked motor deficits (Shimony et al., 2008) and has the potential to show a relationship between neuroanatomical structure and function (Feys et al., 2010). CP has multiple classification systems due to its heterogeneity (Bax et al., 2005). Using classification subcategories helps clinicians to build a better picture of the motor disorder which is important when comparing, predicting and evaluating any changes that occur over time. The traditional classification system for CP is based on topographical classification of motor impairment (the involvement of limbs, trunk and oropharynx). This classification focuses principally on the distributional pattern of the affected limbs (quadriplegia, diplegia, hemiplegia, monoplegia and triplegia) (Bialik and Givon, 2009). Recently, a more comprehensive classification system (Bax et al., 2005; Bialik and Givon, 2009) has been proposed to obtain a complete picture of CP and is based on several components, including motor abnormalities, associated abnormalities, topographical distribution and neuroanatomical findings. Other motor assessments for young children include the Alberta Infant Motor Scale (AIMS), an observational assessment scale constructed to measure gross motor maturation in infants 1 month to the age of independent walking. Based upon the literature, 58 items were generated and organized into four positions: prone, supine, sitting and standing. Each item describes three aspects of motor performance—weight-bearing, posture and antigravity movements. The scale is unidimensional, and measures the construct of motor maturation (Piper et al., 1992, Piper and Darrah, 1994). The score for each position is summed to obtain a total raw score, then converted to an age-based percentile rank based on Canadian normative data. The AIMS has a high degree of test-retest, intra-rater and inter-rater reliability when it is used to measure typically developing infants (Cui et al., 2009; Jeng et al., 2000; Pin et al., 2010; Snyder et al., 2008). The Bayley Scales of Infant Development, second version (BSID-II) was developed to assess the development of children between 1 and 42 months, and to discriminate typically from non-typically developing children. The BSID-II contains a motor scale (111 items, motor items), a mental scale (178 items) and a behavioural rating scale (30 items) (Bayley, 1993). Topographical classification is based on the parts of the body affected by motor impairments or limitations. Neuroanatomical findings evident on computed tomography or magnetic resonance imaging include ventricular enlargement, white matter loss, or brain anomaly. Recently developed functional neuroimaging tools now make it possible to study non-invasively several aspects of human brain functional reorganization in response to injury (Chugani et al., 1996; Maegaki et al., 1999; Staudt, 2010) (see Chapters 2 and 3). In terms of daily living activities, children with motor impairments have less diversity and a slower rhythm in carrying out activities, with more time needed in activities and personal care (Beckung and Hagberg, 2002; Brown and Gordon, 1987; Ostensjo et al., 2004). Consequently, their participation in social engagements, active recreation, household tasks and activities away from home is reduced (Brown and Gordon, 1987). Although the activity and participation domains are more immediately relevant to the children and their parents, understanding CP at the impairment level clarifies whether an intervention is likely to be effective (Burridge et al., 2009) and to what extent the impairment, or combination of impairments, will limit activity in children with CP. Upper motor neuron lesions, for example, those that occur in utero and in the perinatal period in infants with a potential to develop hemiplegia or diplegia, are categorized as having either positive or negative features (Barnes and Johnson, 2008; Kerr and Selber, 2003; Pandyan et al., 2005). The positive features, ‘signs of presence’, include increased reflex activity (hyperreflexia). The negative features, ‘signs of absence’, are weakness, delayed initiation or failure of muscle activation, reduced dexterity and co-ordination (i.e., reduced selective or task-specific motor control), sensory deficits and fatigue. Associated with these features are adaptive or secondary sequelae that occur over time. Some of the later-onset features relate to non-neurogenic, mechanical changes that develop as a consequence of the primary impairments (Turk et al., 2008) together with habitual patterns of active limb use or limb disuse. Morphological and mechanical changes include changes to muscle and tendon length (e.g., contractures) and changes in the mechanical properties of muscles and in connective tissues (Burridge et al., 2009; Mayer, 1997). Although the lesion that occurs in the developing brain is considered to be non-progressive, the secondary musculoskeletal pathology progresses over time (Boyd and Graham, 1997), as children grow and develop. Currently we understand impairments and adaptations to be interrelated, at least in their cause and presentation, with activity limitations, but an understanding of which are the primary contributors to functional motor deficits is still in its infancy. Nevertheless, it seems certain that the neuromotor impairments with the greatest impact are decreased or impaired activation of muscles resulting in weakness and poor motor control/co-ordination. Clinicians have tended to concentrate on novel features of the upper motor neuron syndrome such as spasticity and have neglected to some extent the abnormalities of muscle activation and motor control together with the secondary or adaptive features. However, it is these combined features that lead to activity restrictions such as difficulty learning to walk and carrying out other activities of daily living (Turk et al., 2008). The focus in neurorehabilitation for both adults with stroke and children with CP is shifting to the lack of muscle activation, poor motor control, the subsequent loss of mobility, and their relation to the adaptive sequelae and function (Ada et al., 2006; Carr and Shepherd, 2003; Vaz et al., 2006). Muscle weakness has been defined as a difficulty in generating and controlling the necessary voluntary muscle force for effective motor performance (Carr and Shepherd, 1998). Marked strength deficit is a major negative feature and has been well documented in children with CP compared to typically developing children (Damiano et al., 1995b; Eek and Beckung, 2008; Elder et al., 2003; Rose and McGill, 2005; Stackhouse et al., 2005; Wiley and Damiano, 1998). Inability to voluntarily activate the muscle to the appropriate level (i.e., to generate sufficient muscle force) is believed to be due to several different factors. Among those are loss of motor unit activation, changes in recruitment ordering, or changes in firing rates of agonist muscles, co-activation of antagonist muscles as well as morphological and mechanical changes in the muscles due to decreased physical activity or disuse (Damiano et al., 2000; Rose and McGill, 1998, 2005; Shepherd, 2001; Tedroff et al., 2008). In addition, muscle weakness may occur as a side effect of some pharmacological interventions such as botulinum toxin (BoNT-A) (Edgar, 2001; Saguil, 2005), although the latter is not typically given to infants under 2 years. Spasticity is said to be the most common form of hypertonicity that affects the majority of children with CP (Sanger et al., 2003). It is defined as ‘a motor disorder characterized by a velocity-dependent increase in tonic stretch reflexes with exaggerated tendon jerks, resulting from hyperexcitability of the stretch reflex, as one component of the upper motor neuron syndrome’ (Lance, 1980). Upper motor neuron syndrome is a clinical term used to indicate disruption of the corticospinal tract at any point along its course and may result in predominantly spastic types. The corticospinal tract descends through the corona radiata and internal capsule to reach the brainstem, midbrain, pons, then the medulla oblongata, and here it forms the pyramid (Filloux, 1996; Fitzgerald, 1992; Priori et al., 2006) and descends in the spinal cord. The term spasticity is used to describe several different features and its use is frequently confusing. Typical tests of spasticity involve an examination of the muscle response to passive movement. Sanger et al. (2003) defined spasticity in terms of the features of the clinical examination as hypertonia in which one or both of the following signs are present: (1) resistance to externally imposed movement increases with increasing speed of stretch and varies with the direction of joint movement; and/or (2) resistance to externally imposed movement rises rapidly above a threshold speed or joint angle. Under the first criterion, an increase in stretch velocity does not mean a more exaggerated response to passive stretch (Powers et al., 1989). However, the resistance to passive movement must be different for high versus low speeds (Sanger et al., 2003). The II (secondary) and Ia (primary) sensory neurons of the muscle spindle provide signals to the CNS about change in muscle length (tonic component) and the rate of change in length (phasic component), respectively. The synaptic connection between the Ia and II neurons with the alpha-motor neurone leads to stretch reflex activation (Matthew 1972 cited in Salazar-Torres et al., 2004). However, until now our understanding of spasticity is limited because spasticity is caused by several mechanisms which are more or less linked and associated with different clinical manifestations (Sindou, 2003). From Lance’s widely used definition, it is evident that the clinical symptom of spasticity is a velocity-dependent increase in tonic stretch reflexes at rest in response to passive movement. Gracies (2005b) simplified the wording of Lance’s definition and presented it as ‘a velocity-dependent increase in reflexes to phasic stretch, in the absence of volitional activity’(see Chapter 5). This definition emphasizes the exaggerated stretch reflex or hyperreflexia (triggered by phasic muscle stretch) as the prominent clinical sign of spasticity (Poon and Hui-Chan, 2009). The stretch reflex has a phasic and a tonic component (Alter, 2004). The phasic response is an initial burst of action potentials that results in a rapid rise of muscle tension, proportional to the velocity of the stretch. The tonic response is a later phase of slow (low-frequency) firing, lasting for the duration of the stretch, which is proportional to the amount of stretch. In the past, spasticity was the focus of research and it was assumed that it can interfere with movement and positioning, contribute to the formation of contractures and musculoskeletal deformities over time, and be a source of discomfort by negatively impacting function, making caregiver tasks, such as transfers and dressing, more difficult (Cardoso et al., 2006; Koman et al., 2004; Petrillo and Knoploch, 1988; Suputtitada, 2000; Voerman et al., 2007). However, contracture is not necessarily linked to spasticity and reduction of spasticity may not prevent deformity. Tedroff et al. (2009) report the outcome of a prospective uncontrolled study on the long-term effect of BoNT-A on the lower limb muscles of children with pyramidal CP. The study group is heterogeneous in terms of age (median 5 years 4 months, range 11 months to 17 years 8 months), GMFCS level (29% level I, 15% level II, 16% level III, 17% level IV, 23% level V), and topographical pattern (50% diplegic CP, 22% hemiplegic CP, 25% tetraplegic CP, 3% dyskinetic CP). Outcome measures were limited to the Modified Ashworth Scale (MAS) and joint range-of-motion measures to estimate muscle ‘tone’, made at a minimum before and 3 months after each injection. Injections were repeated, to a maximum of eight injections per muscle. They found a persistent reduction in muscle tone in the muscles injected but a progressive reduction in joint passive ranges, following an initial improvement. They found that BoNT-A can be effective in reducing muscle tone over a longer period, but not in preventing development of contractures in spastic muscles. They concluded that dissociation between the effects on muscle tone and range of motion (ROM) indicates that development of contractures is not coupled to increased muscle tone only, but might be caused by other mechanisms. Until recently, little work had been done on the role of spasticity during active movement (Damiano et al., 2006) although passive stretch and active movement are likely to elicit different manifestations of spasticity (Fleuren et al., 2009). Spasticity is usually tested by reference to clinical measurements made while the patient lies in a relaxed position, although active voluntary movements such as walking are the target of clinical assessment and intervention (Burridge and McLellan, 2000). There are difficulties in studying reflex activity during active movement, possibly due to difficulty in differentiating it from voluntary muscle contraction (Fleuren et al., 2009). Spasticity during passive movement may be greater than, the same as, or less than during active voluntary movement. For this reason it is important not to assume that what is detected during passive examination will necessarily reflect what is happening during functional movements (Burne et al., 2005; Burridge and McLellan, 2000; Dietz, 2000; Fleuren et al., 2009). Stretch reflex hyperexcitability has little functional consequence in spastic patients due to its disappearance during active movement in the agonist muscle although it may be manifest at rest (Ada et al., 1998; Burne et al., 2005; Morita et al., 2001; Nielsen et al., 2007). However, it should be pointed out that the functional significance of spasticity does not solely relate to the agonist muscle, but also to the antagonist muscles (Nielsen et al., 2007). Crenna (1998, 1999) carried out one of the very few investigations of reflex hyperactivity during active movement. He noted that abnormal stretch responses were more easily elicited during lengthening contractions around the time of ground contact of the stride than at other points in the cycle (Crenna, 1998). He attributed this to the floor contact as a critical event that may be more dependent on supraspinal rather than peripheral control, and the disruptions in movement in other parts of the cycle may be attributable to weakness, co-contraction and passive muscle properties, rather than to stretch responses. Dietz and Sinkjaer (2007) showed in their review that oversensitive muscle stretch receptors (hyperreflexia) appear to make a relatively minor contribution to dysfunctional motor performance while secondary changes in mechanical muscle fibre properties have a major role in spastic movement disorder. Clinicians and researchers frequently group the passive mechanical properties together with the neurally mediated phenomena and refer to them as spasticity. It is important to distinguish between reflex- and non-reflex-mediated stiffness in order to understand and reliably evaluate spasticity’s mechanisms because it has consequences for treatment (Zhang et al., 2000). For example, Burridge and McLellan (2000) showed that stroke patients who had poor control of ankle movement and spasticity/reflex hyperactivity were more likely to respond well to functional electrical stimulation (FES), whereas those with mechanical resistance to passive movement and with normal muscle activation respond less well. Resistance to passive movement, however, occurs not only as a result of increased neuromuscular activity but also as a function of the passive mechanical properties of the muscle and its connective tissue content (passive stiffness) (Berger et al., 1984; Hufschmidt and Mauritz, 1985; Johnson, 2002; Sinkjaer et al., 1993). Therefore, these need to be differentiated from each other. Research has shown that morphological modifications of muscle fibres due to disuse or immobilization lead to changes in their neural activation. In animal studies, Giroux et al. (2005) observed a loss of muscle weight and atrophy in both the soleus and peroneus longus of wistar female rats after four weeks of immobilization due to a reduction in mean fibre cross-sectional area (CSA). The morphological modifications were accompanied by an increase in the integrated EMG amplitude. Rosant et al. (2006) related the spindle discharges to stiffness of the muscle–tendon unit using a spindle efficacy index, defined as the ratio between spindle sensitivity and incremental stiffness. They verified that changes in muscle stiffness contribute to changes in spindle responses to stretch after 21 days of hindlimb unloading in rat soleus muscle. There is a possibility that stretch reflex hypersensitivity may be an adaptive response evident sometime after injury and occurring in conjunction with length changes. The increased quantum of connective tissue found after immobilization in a shortened position reduces muscle compliance. This leads to an increase in the spindle response to a given amount of stretching force, as the pull is more efficiently transmitted to the spindles in a less extensible muscle (Gracies, 2005a, 2005b; Maier et al., 1972). Thus, when muscle fibres and spindles become short, sensitivity of the stretch reflex increases. In human studies, although Blackburn et al. (2008) found that soleus stretch reflex latency and amplitude did not differ among individuals with high (men) or low (women) triceps surae stiffness, a significant number of authors were of a different opinion. Kamper et al. (2001) suggested that absolute muscle fibre length plays a highly significant role in the spastic reflex response to imposed movements of the elbow flexors in individuals with chronic spastic hemiplegia following stroke. In a similar way, Meinders et al. (1996) showed that stretch reflex activity varies with ankle position. Although the exact mechanisms responsible for the disuse-induced loss of neuromotor control are far from understood, it does seem that disuse results in reductions in motor performance that are likely related to adaptations in neurophysiologic parameters (Clark, 2009). Passive response to stretch (passive mechanical properties) When a joint is moved passively by an external force to produce displacement, it simply resists the motion by exhibiting a measurable resistance even when skeletal muscles’ motor neurons are quiescent and their myofibres are not actively contracting. This behaviour is called passive muscle stiffness (Schleip et al., 2006). The resistance is derived from the myotendinous unit and its associated connective tissues, but also integrates resistance to motion provided by skin, the joint capsule and ligamentous structures. A muscle–tendon unit (MTU) is a composite structure that includes muscle fibres, the supportive connective tissues within and around the muscle belly, and dense regular connective tissue of the tendons that secure the MTU to bone (Gajdosik and Gajdosik, 2006). According to Hill’s model (Fig. 4.1), these structures are arranged in three separate components and this explains the role of elastic energy in human movement (Hill 1938, cited in Wilson and Flanagan, 2008). They consist of a contractile component (CC), a series elastic component (SEC), and a parallel elastic component (PEC), each of which contributes to overall muscle tension and may be classified as either elastic or viscous (Alter, 2004). Figure 4.1 Hill’s functional muscle model consists of a series elastic component (SEC), a contractile component (CC) and an elastic component (PEC) parallel to the CC. [Adapted with slight modifications from Wilson and Flanagan (2008). The role of elastic energy in activities with high force and power requirements: a brief review. J Strength Cond Res, 22, 1705−1715, with permission from Wolters Kluwer Health.] The cross-bridge component is the active force-generating structure within the myofibrils and is composed of thousands of sarcomeres connected in series. The sarcomere is made of multiple actin and myosin filaments, and with their cross-bridges they produce contraction (Edman, 2003; Givli and Bhattacharya, 2009). The maximum force that can be generated by a muscle fibre or single sarcomere can be affected by the length of muscle, velocity of muscle contraction, type of contraction, frequency of stimulation, motor unit recruitment, and muscle fibre alignment and type (Trew and Everett, 2005). The active curve is obtained only indirectly by subtracting the passive length–tension curve (measured in the absence of active simulation) from the total forces throughout the full length of the muscle (Gajdosik, 2001). The series elastic component is a non-CC of muscle that lies in series with muscle fibres (Hill, 1950). It stores energy when stretched and makes a major contribution to the elasticity of the human skeleton (Hill, 1950). Tendons are the major representatives of the series elastic component, but the cross-bridges between actin and myosin, within the muscle fibre, may also contribute (Herzog, 1997). During passive stretch, the major deformation occurs more in the PEC than in the tendons (Wilson and Flanagan, 2008). The series elastic component’s function is to transmit the forces of the contraction and to store and later release elastic energy (Lindstedt et al., 2002; Roberts, 2002; Wilson and Flanagan, 2008). Ishikawa et al. (2005) suggested that the elastic recoil takes place not as a spring-like bouncing but as a catapult action in natural human walking. It has been shown that the series elastic component adapts its properties according to the functional demand (Almeida-Silveira et al., 2000; Lindstedt et al., 2002; Roberts, 2002). The PEC stores elastic energy in parallel to the CCs of a muscle. Passive elements, such as titin, act in parallel with both the contractile element and the series elastic component, or in parallel with only the contractile element (MacIntosh and MacNaughton, 2005). Increasing the muscle length, and stretching the PEC, increases the passive force or resting tension in muscle (passive force–length relationship) (Alter, 2004). Gajdosik (2001) suggested that the cytoskeleton of the sarcomere and intramuscular connective tissue constitute PECs that contribute to passive tension, modification of which could lead to a change in overall stiffness of the MTU. The structures that contribute the majority of the passive force at the MTU are not entirely known, but it is often assumed that connective tissue, its collagen content, titin filaments and desmin are a source of passive tension if the muscle is stretched beyond its resting length (Bensamoun et al., 2006; Gajdosik and Gajdosik, 2006; Lindstedt et al., 2002; Wang et al., 1993). The connective tissues of muscle, divided into endomysium, perimysium and epimysium, are usually considered to play a major role in the development of passive tension while not actively generating force (Gajdosik, 2001). The endomysium surrounds an individual muscle fibre (cell), the perimysium encircles a group of muscle fibres forming a fascicle, and the epimysium encircles all the fascicles to form the complete muscle (Purslow, 1989). The ensemble of these tissue sheaths, called the fascia, encloses the muscles and ultimately connects them at their ends to the tendons (Van Loocke, 2007). Overstretching of the muscle fibre bundles is prevented by the perimysium and is thought to be the major contributor to passive muscle resistance due to its relative abundance within all components of the connectives tissues (Gajdosik, 2001; Purslow, 1989). The passive mechanical properties of muscle are dependent on the amount, type and architectural organization of collagen fibrils (Mockford and Caulton, 2010). Booth et al. (2001) have shown that collagen is increased in the spastic muscles of children with CP and that the amount of total collagen correlates with the severity of their disorder. The distribution of this increased collagen is consistent with it playing a role in increased muscle stiffness, and collagen content in muscle can change in response to altered use patterns (Herbert, 1988; Waterman-Storer, 1991) or pathologies (Booth et al., 2001). Among the intracellular proteins, which include titin, nebulin, desmin, troponin and tropomyosin, titin and desmin are the most important contributors to the resistive force during passive muscle stretching (Balogh et al., 2005; Gajdosik and Gajdosik, 2006; Prado et al., 2005). The giant titin molecules’ protein (also called connectin) is a subcellular component macromolecule that connects the ends of the myosin filament to the Z-disk and overlaps in the Z-disk and M-band of the sarcomere, forming a continuous elastic filament system inside the myofibre (Craig and Padrón, 2004; Maruyama and Kimura, 2000; Skeie, 2000). Consequently, titin is frequently assumed to be the main structural element responsible for the even distribution of sarcomeric length in non-activated muscle (Goulding et al., 1997; MacIntosh and MacNaughton, 2005). Strain of the titin protein of the endosarcomeric cytoskeleton and of the desmin protein of the exosarcomeric cytoskeleton within the muscle fibre contribute to increased passive resistance during passive lengthening (Bartoo et al., 1997; Gajdosik et al., 2005; Wang et al., 1993). Anderson (2002) suggested that the presence of desmin is crucial and titin is unlikely to be responsible for the large increase in passive stiffness observed in whole soleus muscles when desmin is lacking. Desmin (also called skeletin) is an intermediate-size protein of the exosarcomeric cytoskeleton oriented in both the transverse and longitudinal planes of the muscle cell (Tokuyasu et al., 1983; Wang et al., 1993). It serves to interconnect myofibrils at the level of their Z-bands, and to connect Z-bands in series in a single myofibril and in parallel in adjacent myofibrils (Tokuyasu et al., 1983). This arrangement creates a mechanical structural framework of the serial and parallel mechanical connections to form a network for passive force transmission (Boriek et al., 2001; Gajdosik and Gajdosik, 2006). Desmin’s protein will lengthen as the sarcomere is stretched. Hence, it is thought to contribute to the passive muscle stiffness when stretched (Gajdosik and Gajdosik, 2006). Although muscle spasticity is neural in origin, there is strong evidence that spastic muscles are not normal and have an altered intrinsic mechanical structure secondary to spasticity (Foran et al., 2005; Friden and Lieber, 2003; Lieber and Friden, 2002; Smith et al., 2009), together with lack of or altered patterns of use. Sinkjaer and Magnussen (1994) suggested that the reduced maximal voluntary force generation in the spastic limb is not caused by changed contractile properties in the spastic muscles. Friden and Lieber (2003) found that muscle cells obtained from patients with spasticity had a decreased resting sarcomere length and nearly double the elastic modulus of the stress–strain relationship in fibres compared with normal muscle cells. They suggested that dramatic remodelling of intracellular (such as titin) or extracellular (such as collagen) muscle structural components were the structural basis for their observations of increased viscoelastic properties. Details of data demonstrating that titin is actually altered secondary to spasticity are lacking but there is circumstantial evidence to suggest that it is possible (Foran et al., 2005). The passive mechanical properties of isolated muscle cells and small muscle fibre bundles are two structures that may be altered. Lieber et al. (2003) used single fibres and small bundles of muscle cells (5–50 fibres) ensheathed by the connective tissue matrix of the muscle tissue to demonstrate the difference in the mechanical properties of spastic muscle tissue bundles compared with normal muscle fibre bundles. They found that the spastic single fibres were stiffer than the non-spastic single fibres, even though the non-spastic bundles were remarkably stiffer than the spastic bundles. They also found a large amount of poorly organized extracellular material in spastic bundles compared with normal bundles. In addition, in the spastic muscle bundles, 40% of the CSA was occupied by muscle fibres, compared to 95% in normal muscle bundles (Lieber et al., 2004). Malaiya et al. (2007) collected 3D ultrasound images of the medial gastrocnemius muscle belly in 16 children with pyramidal hemiplegic CP (mean age: 7.8 years; range: 4–12 years) and 15 typically developing children (mean age: 9.5 years; range: 4–13 years). They found reduced muscle volume and length of the paretic limb compared to the non-paretic limb without decrease in fascicle length. The authors attributed these changes to the lack of cross-sectional growth. Further investigation is necessary. Mirbagheri et al. (2008) suggested that using the non-paretic limb may not be an appropriate control for the study of neuromuscular properties in hemiparetics because it is not subject to the forces and usage found in a typically developing child. In histological studies, Rose et al. (1994) showed that children with CP have abnormal variation in the size of muscle fibres and altered distribution of fibre types. Changes in muscle architecture are not limited to passive condition only, but also affect the contractile properties of muscle fascicles (Gao and Zhang, 2008). Ito et al. (1996) studied the histopathology of spastic muscles in nine children with pyramidal CP (age range: 6–18 years) using specimens obtained from the gastrocnemius muscles during orthopaedic operations. They found changes in fibre type distribution, i.e., type 1 fibre predominance and type 2B fibre deficiency. In agreement with previous observations, Marbini et al. (2002) performed muscle biopsy on adductor longus (16 cases) and triceps surae muscles (four cases) during tendon lengthening operations for 20 children with pyramidal CP aged from 4 to 16 years old (mean: 9.4 years). They showed mild myopathic changes, type 1 predominance, and type 1 and type 2 hypotrophy. Mohagheghi et al. (2008) found in an in vivo study of 18 children with diplegia (aged 2–15 years) and 50 typically developing children that children with diplegia had shorter fascicle lengths in the gastrocnemius muscle compared to those typically developing, irrespective of whether absolute or normalized (against leg length) fascicle lengths were compared. Shortland et al. (2002) reported architectural variables of the medial gastrocnemius in five normally developing adults (aged 24–36 years) and five typically developing children (aged 7–11years), and in seven children with spastic diplegia (aged 6–13 years) who had plantarflexion contractures of greater than 10° with the knee extended. They showed a dependence of deep fascicle angle (increasing with increasing plantarflexion) and fascicle length (decreasing with increasing plantarflexion) on ankle joint angle. However, the architecture of the medial gastrocnemius (fascicle length) in normally developing children and children with spastic diplegia does not differ greatly at a common angle of 30° of plantarflexion or at the resting ankle angles.
Functional effects of neural impairments and subsequent adaptations
Introduction
Prevalence and aetiology
Classification of cerebral palsy
Motor abnormalities
 The nature and type of the motor disorder: CP can be divided into two major physiologic classifications (Jones et al., 2007)—pyramidal and extrapyramidal, indicating the area of the brain that has been affected as well as the resulting predominant motor disorder. Pyramidal CP is the common subtype (Andersen et al., 2008; Himmelmann et al., 2006, 2005; Howard et al., 2005; Jain et al., 2008; McClelland et al., 2006) and results from defects or damage to the corticospinal pathways in the brain, also described as upper motor neuron damage. It is commonly associated with muscle weakness and loss of motor control, with spasticity illustrated by hyperreflexia, clonus, and extensor Babinski response. Extrapyramidal CP is caused by damage to nerve cells/neural pathways outside the pyramidal tracts in the basal ganglia or the cerebellum. It is typically divided into two subtypes, dyskinetic and ataxic.
The nature and type of the motor disorder: CP can be divided into two major physiologic classifications (Jones et al., 2007)—pyramidal and extrapyramidal, indicating the area of the brain that has been affected as well as the resulting predominant motor disorder. Pyramidal CP is the common subtype (Andersen et al., 2008; Himmelmann et al., 2006, 2005; Howard et al., 2005; Jain et al., 2008; McClelland et al., 2006) and results from defects or damage to the corticospinal pathways in the brain, also described as upper motor neuron damage. It is commonly associated with muscle weakness and loss of motor control, with spasticity illustrated by hyperreflexia, clonus, and extensor Babinski response. Extrapyramidal CP is caused by damage to nerve cells/neural pathways outside the pyramidal tracts in the basal ganglia or the cerebellum. It is typically divided into two subtypes, dyskinetic and ataxic.
 Functional motor abilities: Evaluating the functional consequences of CP is the key to treating children with CP. Several scales have been developed to measure function in all body areas. They include the Gross Motor Function Classification System (GMFCS), which is often used to evaluate children with CP and is internationally validated in relation to ambulation and activity limitations (Hanna et al., 2009; Palisano et al., 1997, 2006; Pfeifer et al., 2009; Rosenbaum et al., 2008). It is a five-level, age-categorized system developed to classify the severity of motor involvement in children with CP and is based on functional abilities and limitations. The scale has a section on infants that can be used to test those who are younger than 2 years of age. They can be given a provisional score until they are reviewed at 2 years old, as more clinical information becomes available (Gorter et al., 2009b). Infants are classified as level I when they move in and out of sitting and floor sit with both hands free to manipulate objects; they crawl on hands and knees, pull to stand and take steps holding on to furniture; and they walk between 18 months and 2 years of age without the need for any assistive mobility device. Infants at level V have physical impairments limiting voluntary control of movement; they are unable to maintain antigravity head and trunk postures in prone and sitting; and they require an adult’s help to roll.
Functional motor abilities: Evaluating the functional consequences of CP is the key to treating children with CP. Several scales have been developed to measure function in all body areas. They include the Gross Motor Function Classification System (GMFCS), which is often used to evaluate children with CP and is internationally validated in relation to ambulation and activity limitations (Hanna et al., 2009; Palisano et al., 1997, 2006; Pfeifer et al., 2009; Rosenbaum et al., 2008). It is a five-level, age-categorized system developed to classify the severity of motor involvement in children with CP and is based on functional abilities and limitations. The scale has a section on infants that can be used to test those who are younger than 2 years of age. They can be given a provisional score until they are reviewed at 2 years old, as more clinical information becomes available (Gorter et al., 2009b). Infants are classified as level I when they move in and out of sitting and floor sit with both hands free to manipulate objects; they crawl on hands and knees, pull to stand and take steps holding on to furniture; and they walk between 18 months and 2 years of age without the need for any assistive mobility device. Infants at level V have physical impairments limiting voluntary control of movement; they are unable to maintain antigravity head and trunk postures in prone and sitting; and they require an adult’s help to roll.
Topographical and neuroanatomical findings
Motor impairments and muscle adaptations
Impaired activation of motor neurons and poor motor control
Spasticity (reflexive response to stretch)
Differentiation between neural (stretch reflex hypersensitivity) and mechanical factors
Changes in reflex sensitivity as contracture develops
Adaptive changes in passive mechanical properties of muscle
Muscle mechanical components
Sources of passive tension or stiffness
Related posts:
 Re-thinking the brain: new insights into early experience and brain development
Effects of motor activity on brain and muscle development in cerebral palsy
B: Findings from a series of studies of passive mechanical properties of muscle in young children with cerebral palsy
Treadmill training in early infancy: sensory and motor effects
Very early upper limb interventions for infants with asymmetric brain lesions
Training lower limb performance in early infancy: Support, balance and propulsion
Re-thinking the brain: new insights into early experience and brain development
Effects of motor activity on brain and muscle development in cerebral palsy
B: Findings from a series of studies of passive mechanical properties of muscle in young children with cerebral palsy
Treadmill training in early infancy: sensory and motor effects
Very early upper limb interventions for infants with asymmetric brain lesions
Training lower limb performance in early infancy: Support, balance and propulsion
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Functional effects of neural impairments and subsequent adaptations
