Metabolic myopathies are a group of genetic disorders specifically affecting glucose/glycogen, lipid, and mitochondrial metabolism. The main metabolic myopathies that are evaluated in this article are the mitochondrial myopathies, fatty acid oxidation defects, and glycogen storage disease. This article focuses on the usefulness of exercise in the evaluation of genetic metabolic myopathies.
Overview of metabolic myopathies
Metabolic myopathies are a group of genetic disorders specifically affecting glucose/glycogen, lipid, and mitochondrial metabolism. The main metabolic myopathies that are evaluated in this article are the mitochondrial myopathies, fatty acid oxidation defects (FAOD), and glycogen storage disease (GSD). Traditionally, defects in the enzyme myoadenylate deaminase (AMPD1) were considered to be a common metabolic myopathy; however, it is unlikely that AMPD1 deficiency is a true metabolic myopathy given the high prevalence in the general population (2%), that otherwise asymptomatic people (even athletes) can have this mutation with no exercise intolerance, and that high-intensity exercise does not alter energy status in skeletal muscle of patients homozygous for the common AMPD1 mutation. A summary of some of the more common metabolic myopathies is presented in Table 1 .
| Mitochondrial myopathy | MELAS syndrome, complex IV deficiency, cytochrome b mutations, complex I deficiency, Kearn-Syre syndrome, chronic progressive external ophthalmoplegia |
| Fat oxidation defect | CPT2 deficiency, trifunctional protein deficiency, VLCAD deficiency, glutaric aciduria type 2 |
| GSD | Myophosphorylase deficiency (McArdle disease), PFK deficiency (Tarui disease), phosphorylase b kinase deficiency, lactate dehydrogenase deficiency |
There are also medications that can interfere with intermediary metabolism and unmask or exacerbate an underlying genetic metabolic myopathy. The most common example of this phenomenon is 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor (statins) medications leading to rhabdomyolysis and occurring in about 0.1% of all patients taking statins. The cause of statin-associated myopathies is unclear; however, the depletion of coenzyme Q10 may be a more relevant mechanism in the rare cases when a patient with a genetic metabolic myopathy is prescribed a statin. It is also known that patients taking statin medications are more susceptible to exercise-induced muscle damage with an increased serum creatine kinase level (hyperCKemia), and this must be considered in the evaluation of a patient with exercise-induced muscle cramps/myalgias and hyperCKemia. Statin myopathies are classified as toxic myopathies and are not considered further in this article, which focuses on the usefulness of exercise in the evaluation of genetic metabolic myopathies.
The metabolic responses to exercise
The specific metabolic fuel supplied to muscle is a function of the intensity and duration of exercise. At the onset of muscle contraction, there is the hydrolysis of adenosine triphosphate (ATP) to drive the various ATPases in muscle (myosin ATPase, calcium ATPase, Na + /K + ATPase). The alteration in cellular energy charge leads initially to the activation of the anaerobic pathways through a variety of mechanisms via mediators such as inorganic phosphate, protons, adenosine monophosphate (AMP), and calcium. Instantaneously, the accumulation of adenosine diphosphate (ADP) in muscle results in ATP generation through the adenylate kinase and AMP deaminase system, with flux through this pathway ultimately leading to the production of ammonia and uric acid ( Box 1 ). Within the first couple of seconds of contraction, there is also rephosphorylation of ADP to ATP through phosphocreatine stores catalyzed by the enzyme creatine kinase. The energy-buffering action of phosphocreatine is a function of muscle phosphocreatine stores, which can usually provide energy for close to 10 seconds of sustained muscle contraction. Within the first seconds of contraction, there is also the activation of glycogenolysis by phosphorylation of myophosphorylase (activated form) and glycolysis through phosphofructokinase (PFK) activation by allosteric regulation. In anaerobic conditions, the glycolytic pathway leads to the accumulation of lactate and protons that can eventually inhibit muscle contraction and limit the duration of activities supported solely by anaerobic glycolysis/glycogenolysis (see Box 1 ).
- 1.
Adenylate kinase/myoadenylate deaminase
- a.
ATP>( ATPase )>ADP+ADP>( AK )>ATP+AMP>( AMPD1 )>IMP+NH 3 >>( XO )>uric acid
- a.
- 2.
Phosphocreatine/creatine temporal energy buffering
- a.
ATP>( ATPase )>ADP+PCr+H + >( cCK )>Cr+ATP
- a.
- 3.
Anaerobic glycogenolysis/glycolysis
- a.
Glycogen>( myophosphorylase )>glucose-6-phosphate (see glycolysis)
- b.
Glucose>( hexokinase )>glucose-6-phosphate+ATP>fructose-6-phosphate+ADP>( PFK )>fructose1,6-bisphosphate>glyceraldehyde-3-phosphate+NAD>1,3-bisphosphoglycerate+NADH+H + +ADP>3-phosphoglycerate+ATP⋙phosphoenylpyruvate+ADP>pyruvate+ATP+NADH+H + >( LDH )>lactate+NADH
- a.
Note: the key enzymes are in italics.
Abbreviations: AK, adenylate kinase; AMPD1, myoadenylate deaminase; ATP, adenosine triphosphate; cCK, cytosolic creatine kinase; LDH, lactate dehydrogenase; NAD, nicotinamide adenine dinucleotide; PFK, phosphofructokinase; XO, xanthine oxidase.
Even with lower intensity activity, the first seconds to minutes of muscle contraction are relatively anaerobic; however, as capillaries dilate and deliver more oxygen to the working muscle there is a progressive increase in delivery of oxygen and activation of oxidative metabolism. The creatine kinase system leads to an increase in ADP in the mitochondrial matrix that further activates mitochondrial respiration (a form of state 3 respiration). The contraction-induced increase in intracellular calcium activates the tricarboxylic acid cycle; the increase in AMP also leads to activation of AMP-dependent kinase (AMPK), which phosphorylates several rate-limiting enzymes involved in the regulation of aerobic metabolism.
The final common pathway for aerobic metabolism is the mitochondria, where reducing equivalents (NADH + H + and FADH 2 ) are delivered to complex I and II respectively, from the decarboxylation of amino acids, free fatty acids (FFAs), and carbohydrate (glucose). Most of the glucose for glycolysis comes from muscle glycogen; however, some is supplied from blood glucose derived from hepatic glycogenolysis or gluconeogenesis. FFAs are derived from adipocyte-derived triacylglycerol lipolysis (peripheral lipolysis) and from the intramuscular hydrolysis of intramyocellular lipids (IMCL). Several amino acids can be oxidized in exercising skeletal muscle; however, at most they contribute 5% to the total energy requirements and usually under conditions of glycogen depletion. In addition, there are no known disorders of amino acid metabolism that lead to an exercise-induced metabolic myopathy, with most leading to central nervous system disorders with developmental delay, regression, and seizures.
Exercise intensity during endurance exercise is measured as oxygen consumption (V o 2 ). At the cellular level, V o 2 is a function of cardiac output (heart rate [HR] × stroke volume) and the peripheral oxygen extraction by the tissues (arterial−venous oxygen content of blood). V o 2 is measured during exercise testing at the whole body level using a metabolic cart that consists of oxygen and carbon dioxide sensors and a pneumotachometer to measure ventilation. V o 2 reflects the total oxygen consumption over unit time calculated from the difference between room oxygen (20.93%) and expired gas as a percentage, multiplied by the minute ventilation in liters per minute. Carbon dioxide sensors are also included in the metabolic cart to measure the volume of carbon dioxide production (V co 2 ); the ratio of the V co 2 /V o 2 is called the respiratory exchange ratio (RER), which reflects the type of fuel being consumed during steady state exercise ( Fig. 1 ). During the maximal V o 2 (V o 2max ) exercise test, the intensity (load) and/or speed (in the case of a treadmill) are progressively increased until the patient reaches maximal capacity and V o 2 plateaus. During this progression, there is a gradual increase in reliance on anaerobic energy systems, with an exponential increase in respiration and lactate production (anaerobic/lactate threshold); the RER will exceed unity and is usually more than 1.12 at V o 2max .
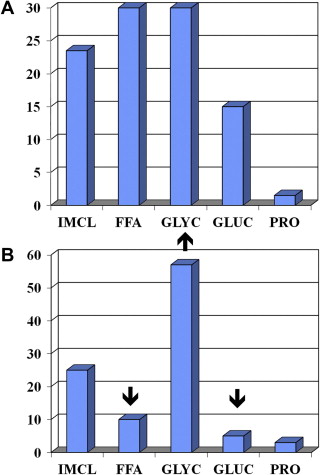
At low exercise intensities (∼35% V o 2max ), energy supply is derived from peripheral lipolysis, IMCL hydrolysis, and plasma glucose, with minimal contribution from muscle glycogen (see Fig. 1 ). At an exercise intensity of ∼65% of V o 2max , there is proportionately greater contribution from IMCL and glycogen, and at 85% of V o 2max there is a proportionately greater contribution to the energy supply from intramuscular glycogen stores. For top sport runners, most of the energy supply to run a marathon comes from muscle glycogenolysis. During fixed lower intensity exercise for a long duration (ie, 2-hour cycle at 65% of V o 2max ), there is also a progressive increase in the contribution from FFAs (IMCL and plasma FFA) as muscle glycogen stores begin to deplete (see Fig. 1 ).
The metabolic responses to exercise
The specific metabolic fuel supplied to muscle is a function of the intensity and duration of exercise. At the onset of muscle contraction, there is the hydrolysis of adenosine triphosphate (ATP) to drive the various ATPases in muscle (myosin ATPase, calcium ATPase, Na + /K + ATPase). The alteration in cellular energy charge leads initially to the activation of the anaerobic pathways through a variety of mechanisms via mediators such as inorganic phosphate, protons, adenosine monophosphate (AMP), and calcium. Instantaneously, the accumulation of adenosine diphosphate (ADP) in muscle results in ATP generation through the adenylate kinase and AMP deaminase system, with flux through this pathway ultimately leading to the production of ammonia and uric acid ( Box 1 ). Within the first couple of seconds of contraction, there is also rephosphorylation of ADP to ATP through phosphocreatine stores catalyzed by the enzyme creatine kinase. The energy-buffering action of phosphocreatine is a function of muscle phosphocreatine stores, which can usually provide energy for close to 10 seconds of sustained muscle contraction. Within the first seconds of contraction, there is also the activation of glycogenolysis by phosphorylation of myophosphorylase (activated form) and glycolysis through phosphofructokinase (PFK) activation by allosteric regulation. In anaerobic conditions, the glycolytic pathway leads to the accumulation of lactate and protons that can eventually inhibit muscle contraction and limit the duration of activities supported solely by anaerobic glycolysis/glycogenolysis (see Box 1 ).
- 1.
Adenylate kinase/myoadenylate deaminase
- a.
ATP>( ATPase )>ADP+ADP>( AK )>ATP+AMP>( AMPD1 )>IMP+NH 3 >>( XO )>uric acid
- a.
- 2.
Phosphocreatine/creatine temporal energy buffering
- a.
ATP>( ATPase )>ADP+PCr+H + >( cCK )>Cr+ATP
- a.
- 3.
Anaerobic glycogenolysis/glycolysis
- a.
Glycogen>( myophosphorylase )>glucose-6-phosphate (see glycolysis)
- b.
Glucose>( hexokinase )>glucose-6-phosphate+ATP>fructose-6-phosphate+ADP>( PFK )>fructose1,6-bisphosphate>glyceraldehyde-3-phosphate+NAD>1,3-bisphosphoglycerate+NADH+H + +ADP>3-phosphoglycerate+ATP⋙phosphoenylpyruvate+ADP>pyruvate+ATP+NADH+H + >( LDH )>lactate+NADH
- a.
Note: the key enzymes are in italics.
Abbreviations: AK, adenylate kinase; AMPD1, myoadenylate deaminase; ATP, adenosine triphosphate; cCK, cytosolic creatine kinase; LDH, lactate dehydrogenase; NAD, nicotinamide adenine dinucleotide; PFK, phosphofructokinase; XO, xanthine oxidase.
Even with lower intensity activity, the first seconds to minutes of muscle contraction are relatively anaerobic; however, as capillaries dilate and deliver more oxygen to the working muscle there is a progressive increase in delivery of oxygen and activation of oxidative metabolism. The creatine kinase system leads to an increase in ADP in the mitochondrial matrix that further activates mitochondrial respiration (a form of state 3 respiration). The contraction-induced increase in intracellular calcium activates the tricarboxylic acid cycle; the increase in AMP also leads to activation of AMP-dependent kinase (AMPK), which phosphorylates several rate-limiting enzymes involved in the regulation of aerobic metabolism.
The final common pathway for aerobic metabolism is the mitochondria, where reducing equivalents (NADH + H + and FADH 2 ) are delivered to complex I and II respectively, from the decarboxylation of amino acids, free fatty acids (FFAs), and carbohydrate (glucose). Most of the glucose for glycolysis comes from muscle glycogen; however, some is supplied from blood glucose derived from hepatic glycogenolysis or gluconeogenesis. FFAs are derived from adipocyte-derived triacylglycerol lipolysis (peripheral lipolysis) and from the intramuscular hydrolysis of intramyocellular lipids (IMCL). Several amino acids can be oxidized in exercising skeletal muscle; however, at most they contribute 5% to the total energy requirements and usually under conditions of glycogen depletion. In addition, there are no known disorders of amino acid metabolism that lead to an exercise-induced metabolic myopathy, with most leading to central nervous system disorders with developmental delay, regression, and seizures.
Exercise intensity during endurance exercise is measured as oxygen consumption (V o 2 ). At the cellular level, V o 2 is a function of cardiac output (heart rate [HR] × stroke volume) and the peripheral oxygen extraction by the tissues (arterial−venous oxygen content of blood). V o 2 is measured during exercise testing at the whole body level using a metabolic cart that consists of oxygen and carbon dioxide sensors and a pneumotachometer to measure ventilation. V o 2 reflects the total oxygen consumption over unit time calculated from the difference between room oxygen (20.93%) and expired gas as a percentage, multiplied by the minute ventilation in liters per minute. Carbon dioxide sensors are also included in the metabolic cart to measure the volume of carbon dioxide production (V co 2 ); the ratio of the V co 2 /V o 2 is called the respiratory exchange ratio (RER), which reflects the type of fuel being consumed during steady state exercise ( Fig. 1 ). During the maximal V o 2 (V o 2max ) exercise test, the intensity (load) and/or speed (in the case of a treadmill) are progressively increased until the patient reaches maximal capacity and V o 2 plateaus. During this progression, there is a gradual increase in reliance on anaerobic energy systems, with an exponential increase in respiration and lactate production (anaerobic/lactate threshold); the RER will exceed unity and is usually more than 1.12 at V o 2max .
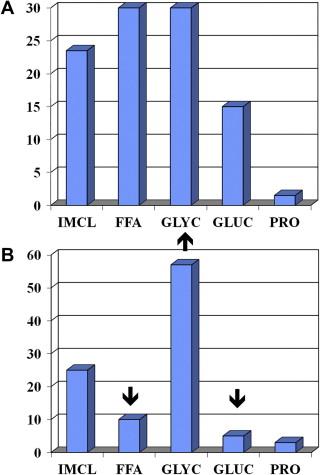
At low exercise intensities (∼35% V o 2max ), energy supply is derived from peripheral lipolysis, IMCL hydrolysis, and plasma glucose, with minimal contribution from muscle glycogen (see Fig. 1 ). At an exercise intensity of ∼65% of V o 2max , there is proportionately greater contribution from IMCL and glycogen, and at 85% of V o 2max there is a proportionately greater contribution to the energy supply from intramuscular glycogen stores. For top sport runners, most of the energy supply to run a marathon comes from muscle glycogenolysis. During fixed lower intensity exercise for a long duration (ie, 2-hour cycle at 65% of V o 2max ), there is also a progressive increase in the contribution from FFAs (IMCL and plasma FFA) as muscle glycogen stores begin to deplete (see Fig. 1 ).
Related posts:
 Advancing Neuromuscular Medicine Through Integration, Translation, and Collaboration
Advancing Neuromuscular Medicine Through Integration, Translation, and Collaboration
 Patient Safety in Cancer Rehabilitation
Patient Safety in Cancer Rehabilitation
 The Utility of Electromyography and Mechanomyography for Assessing Neuromuscular Function: A Noninvasive Approach
The Utility of Electromyography and Mechanomyography for Assessing Neuromuscular Function: A Noninvasive Approach
 Skeletal Muscle Edema in Muscular Dystrophy: Clinical and Diagnostic Implications
Skeletal Muscle Edema in Muscular Dystrophy: Clinical and Diagnostic Implications
 Cardiac MRI in Muscular Dystrophy: An Overview and Future Directions
Cardiac MRI in Muscular Dystrophy: An Overview and Future Directions
 The Paradox of Muscle Hypertrophy in Muscular Dystrophy
The Paradox of Muscle Hypertrophy in Muscular Dystrophy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


