Clinical and electrical myotonia is caused by a small group of neuromuscular disorders. This article reviews myotonia and its differential diagnosis. The use of electrodiagnostic testing to evaluate the primary myotonic disorders (myotonic dystrophy and the nondystrophic myotonias) is also discussed.
Key points
- •
Clinical or electrical myotonia is caused by a small group of neuromuscular disorders including the dystrophic and nondystrophic myotonias.
- •
Chloride or sodium muscle channelopathies are the causes of myotonia in the dystrophic and nondystrophic myotonic disorders.
- •
Electrodiagnostic techniques, including needle electromyogram examination, short and long exercise testing, can help distinguish among the various myotonic disorders.
Introduction
Clinical and electrical myotonia is caused by a small group of neuromuscular disorders ( Box 1 ). Myotonia is due to increased excitability of the muscle membrane often caused by dysfunction of muscle ion channels. Clinical myotonia is manifest by incomplete relaxation of muscle following either voluntary muscle contraction or direct muscle percussion. At the bedside, myotonia is clinically demonstrable by slowed muscle relaxation during repetitive hand grip and eye closure or by delayed relaxation of a muscle contraction evoked by tapping various muscles such as the thenar eminence or the finger extensors.
Muscular dystrophies:
- •
Myotonic dystrophy type 1 and 2
- •
Myofibrillar myopathies
Muscle channelopathies:
- •
Nondystrophic myotonia (myotonia congenita, paramyotonia congenita, sodium channel myotonia)
- •
Hyperkalemic periodic paralysis
Metabolic myopathy:
- •
Acid maltase deficiency a,
- •
Debrancher deficiency a,
- •
McArdle disease (myophosphorylase deficiency) a,
Toxic myopathies a :
- •
Chloroquine/hydroxychloroquine myopathy
- •
Statin myopathy
- •
Colchicine myopathy
Endocrine myopathies a :
- •
Hypothyroidism
Inflammatory myopathies a,
- •
Polymyositis
- •
Dermatomyositis
a Electrical myotonia without clinical myotonia.
Electrical myotonia is an abnormal spontaneous muscle fiber discharge observed on needle electromyogram (EMG) examination. Electrical myotonia appears as repetitive muscle fiber potential discharges (eg, positive waves or fibrillation potentials) with waxing and waning frequency and amplitude with a firing rate between 20 and 80 Hz ( Fig. 1 A). When played over the audio, myotonic discharges have a characteristic sound of a dive bomber, or in the modern day, an accelerating and decelerating motorcycle engine. Electrical myotonia must be distinguished from neuromyotonia (frequency of greater than 150 Hz with a pinging sound) and chronic repetitive discharges of constant, or less commonly, waning frequency with a machinelike sound.
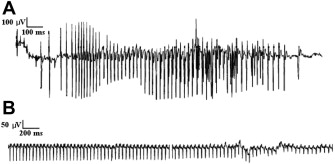
This article focuses on electrodiagnosis of the primary myotonic disorders (myotonic dystrophy and the nondystrophic myotonias [NDMs]) as well as the related periodic paralysis (PP) muscle channelopathies.
Introduction
Clinical and electrical myotonia is caused by a small group of neuromuscular disorders ( Box 1 ). Myotonia is due to increased excitability of the muscle membrane often caused by dysfunction of muscle ion channels. Clinical myotonia is manifest by incomplete relaxation of muscle following either voluntary muscle contraction or direct muscle percussion. At the bedside, myotonia is clinically demonstrable by slowed muscle relaxation during repetitive hand grip and eye closure or by delayed relaxation of a muscle contraction evoked by tapping various muscles such as the thenar eminence or the finger extensors.
Muscular dystrophies:
- •
Myotonic dystrophy type 1 and 2
- •
Myofibrillar myopathies
Muscle channelopathies:
- •
Nondystrophic myotonia (myotonia congenita, paramyotonia congenita, sodium channel myotonia)
- •
Hyperkalemic periodic paralysis
Metabolic myopathy:
- •
Acid maltase deficiency a,
- •
Debrancher deficiency a,
- •
McArdle disease (myophosphorylase deficiency) a,
Toxic myopathies a :
- •
Chloroquine/hydroxychloroquine myopathy
- •
Statin myopathy
- •
Colchicine myopathy
Endocrine myopathies a :
- •
Hypothyroidism
Inflammatory myopathies a,
- •
Polymyositis
- •
Dermatomyositis
a Electrical myotonia without clinical myotonia.
Electrical myotonia is an abnormal spontaneous muscle fiber discharge observed on needle electromyogram (EMG) examination. Electrical myotonia appears as repetitive muscle fiber potential discharges (eg, positive waves or fibrillation potentials) with waxing and waning frequency and amplitude with a firing rate between 20 and 80 Hz ( Fig. 1 A). When played over the audio, myotonic discharges have a characteristic sound of a dive bomber, or in the modern day, an accelerating and decelerating motorcycle engine. Electrical myotonia must be distinguished from neuromyotonia (frequency of greater than 150 Hz with a pinging sound) and chronic repetitive discharges of constant, or less commonly, waning frequency with a machinelike sound.
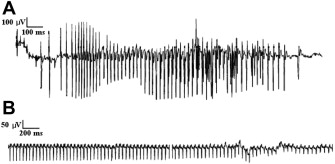
This article focuses on electrodiagnosis of the primary myotonic disorders (myotonic dystrophy and the nondystrophic myotonias [NDMs]) as well as the related periodic paralysis (PP) muscle channelopathies.
Evaluation of channelopathies
Muscle ion channelopathies alter muscle membrane resting potential, resulting in either muscle hyperexcitability (eg, myotonia) or inexcitability (eg, muscle weakness). In the NDMs, muscle membrane hyperexcitability typically results in muscle “stiffness” during voluntary movement because of delayed skeletal muscle relaxation caused by repetitive muscle fiber action potentials (myotonia) as a result of mutations in the chloride (CLCN1) or sodium (SCN4A) skeletal muscle channel genes. By contrast, in the PPs, skeletal muscle membrane, inexcitability results in prolonged episodic muscle weakness caused by mutations in the sodium (SCN4A), calcium (CACN1A), and potassium (KCNJ2) channels. Of note, patients with both hyperkalemic PP and paramyotonia congenita (PC), due to different SCN4A mutations, have myotonia and episodes of skeletal muscle weakness.
Nondystrophic Myotonia
The NDMs include myotonia congenita (MC), PC, and sodium channel myotonia (SCM). The main clinical symptom of NDM is muscle stiffness from myotonia. Some patients also develop weakness and pain. These conditions must be distinguished from myotonic dystrophy type 1 and 2 (DM1 and DM2), which have significant, extramuscular, systemic manifestations. On clinical grounds alone, there is often an overlap among DM2 and NDM. Electrodiagnostic and genetic testing help differentiate among the NDMs and between NDM and DM. When functionally debilitating, myotonia in NDM is treated with sodium channel blockade.
Myotonia congenita
MC presents as either an autosomal recessive form (also known as Becker disease) or a less severe autosomal dominant form (called Thomsen disease.) Both are caused by loss of function mutations in the CLCN-1 chloride channel, resulting in relative depolarization of the muscle membrane. Autosomal recessive MC presents between age 4 and 12 years and autosomal dominant MC presents before age 3 years. Myotonia typically develops with vigorous voluntary movement after resting; a warm-up phenomenon, that is, improvement in muscle relaxation with repetitive hand grip or rarely eye closure is often described. Patients with Becker (but not Thomsen) disease may develop transient weakness or “paresis” lasting seconds to minutes that resolves (like the myotonia) with repetitive muscle contractions.
Paramyotonia congenita (Group 1 SCM)
PC is an autosomal dominant disease due to a gain of function mutation in the SCN4A muscle sodium channel, resulting in relative muscle membrane depolarization. Patients develop not only muscle stiffness due to myotonia but also flaccid muscle weakness when depolarization progresses to membrane inexcitability. Cold temperature and exercise exacerbate myotonia and weakness in this disease. Face and hand muscles are preferentially affected. Most PC patients have paradoxic myotonia of hand grip or eye closure characterized by progressively slower muscle relaxation with repetitive activity, the opposite of the warm-up phenomenon typically seen in the chloride channelopathies. Patients present during the first or second decade. PC is allelic to hyperkalemic PP and like that condition results in prolonged episodes of muscle weakness.
Sodium channel myotonias (Group 2 SCM)
This group of autosomal dominant diseases is also caused by gain of function mutations of the SCN4A channel similar to PC. However, group 2 SCM patients do not typically develop attacks of weakness (as in PC). In contrast to chloride channelopathies, patients with group 2 SCM are often sensitive to potassium, may have eyelid myotonia, lack transient paresis, and more often have pain. SCM can also be distinguished from PC and chloride channelopathies based on electrodiagnostic testing(see later discussion).
Electrodiagnosis of NDM
Commercial genetic testing is available for some of the causative mutations in NDM, but there is a false-negative rate in these conditions as high as 20%. Electrodiagnostic studies are extremely helpful in directing genetic testing and also making the diagnosis of NDM in patients with negative genetic testing. Evidence of membrane hyperexcitability (eg, myotonic discharges) is sought with needle EMG examination, whereas evidence of membrane inexcitability (eg, drop in motor response amplitude) is investigated with long and short exercise testing.
Needle EMG reveals diffuse myotonic discharges in proximal and distal muscles in NDM ; at times the number of myotonic discharges can be so substantial that evaluation of voluntary motor unit potential morphology and recruitment is not possible. In addition to myotonic discharges, low-amplitude (100–600 μV), high-frequency (150–250 Hz) discharges resembling neuromyotonic discharges have recently been observed in some NDM patients. Occasionally, repetitive firing of muscle fibers following a single stimulus termed “postexercise myotonic potentials” can be observed as delayed lower amplitude motor responses following the compound motor action potential (CMAP) during performance of routine nerve conduction studies. Postexercise myotonic potentials are described in both SCN4A and CLCN1 mutations.
In general, the pattern and location of electrical myotonia does not distinguish among the NDM disorders, but the long and especially the short exercise test results can be helpful in this regard. Postexercise recording of serial CMAPs evaluates the functional consequences of ion channel mutations. A long-exercise protocol was initially described, which shows 80% sensitivity in PP and 15% to 30% sensitivity in NDM. Subsequently, a short-exercise protocol was developed that has been shown to be more sensitive for the detection of NDMs in general and useful in differentiating among the individual NDM disorders.
Short-exercise protocol
In the short-exercise protocol, serial CMAPs are recorded from the abductor digiti minimi (ADM) after supramaximal stimulation of the ulnar nerve at the wrist. Supramaximal CMAPs are recorded at baseline and then following 10 seconds of sustained contraction of the ADM. Additional CMAPs are recorded 2 seconds after exercise and then every 10 seconds for a total of 60 seconds; this protocol is repeated 3 times. Postexercise CMAP amplitudes are compared with the patient’s preexercise baseline CMAP amplitudes. The short-exercise protocol is easy to perform and causes minimal patient discomfort when compared with other electrodiagnostic tests such as prolonged repetitive nerve stimulation.
The short-exercise protocol has a high sensitivity of 100% (83%–100%) in PC, 83% (53%–100%) in MC, and 60% in SCM. Repeating the protocol with limb cooling (to 20–25°C) improves the sensitivity in MC (70%–100%) and exaggerates the abnormality observed in PC.
Three patterns are observed in patients with NDM, Fournier I, II, and III. Patients with PC exhibit Fournier pattern I with a decrement in CMAP amplitude (19%–40% less than preexercise baseline CMAP), which persists for 60 seconds and which increases with subsequent trials and with cooling ( Fig. 2 ). This decrement reproduces the weakness in PC patients that develops with exercise, especially with muscle cooling.
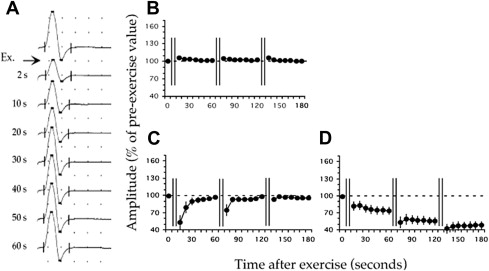
Related posts:
 The Electrodiagnosis of Neuromuscular Disorders
The Electrodiagnosis of Neuromuscular Disorders
 Electrodiagnosis of Cervical Radiculopathy
Electrodiagnosis of Cervical Radiculopathy
 Electrodiagnosis of Brachial Plexopathies and Proximal Upper Extremity Neuropathies
Electrodiagnosis of Lumbar Radiculopathy
An Electrodiagnostic Approach to the Evaluation of Peripheral Neuropathies
Electrodiagnosis of Brachial Plexopathies and Proximal Upper Extremity Neuropathies
Electrodiagnosis of Lumbar Radiculopathy
An Electrodiagnostic Approach to the Evaluation of Peripheral Neuropathies
 Electrodiagnostic Evaluation of Myopathies
Electrodiagnostic Evaluation of Myopathies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


