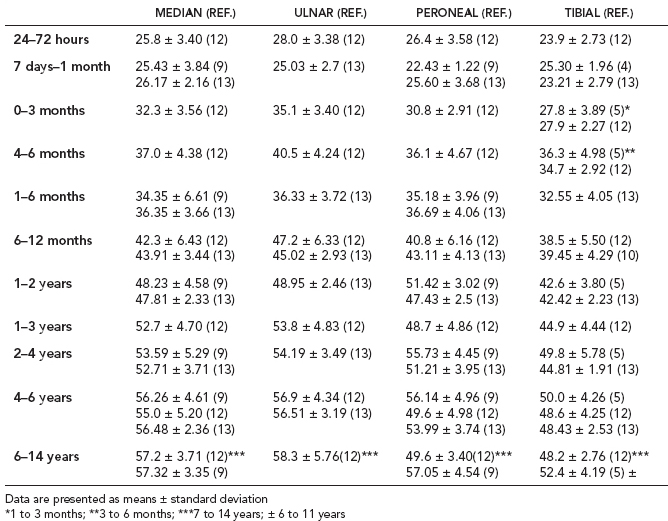
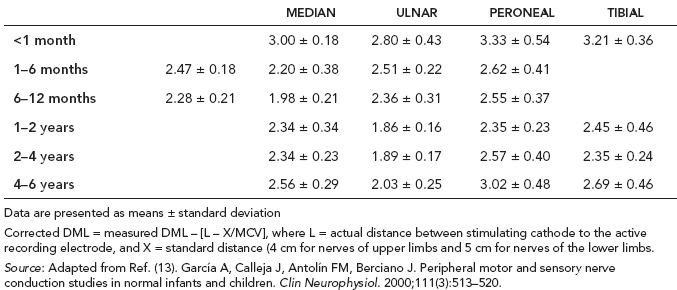
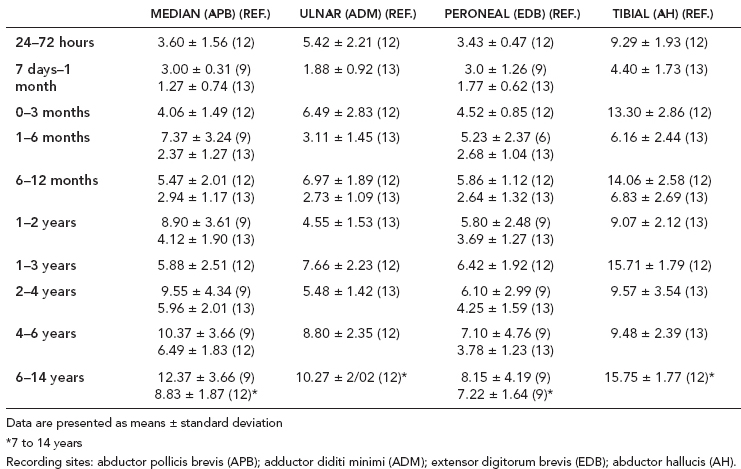
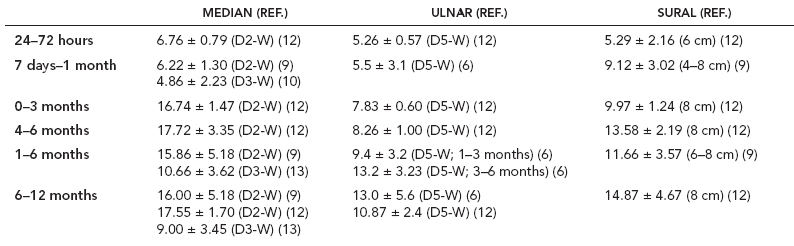
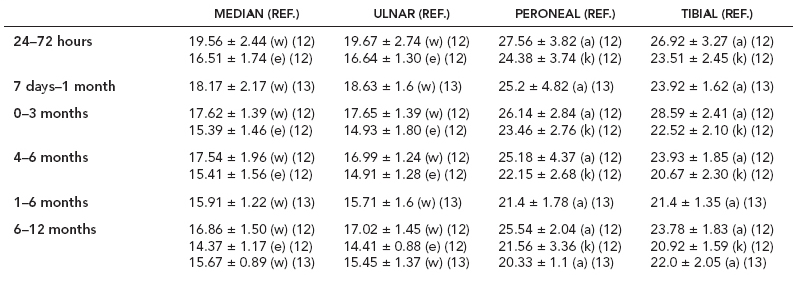
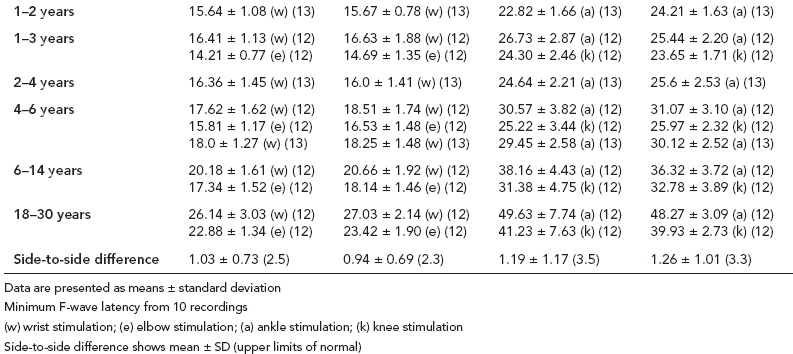
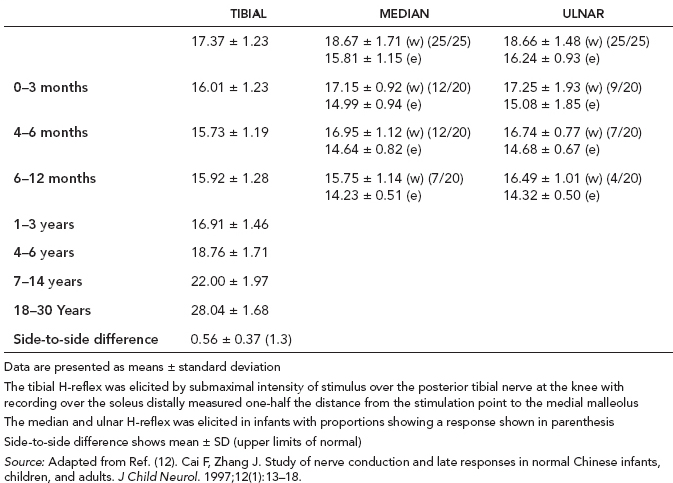
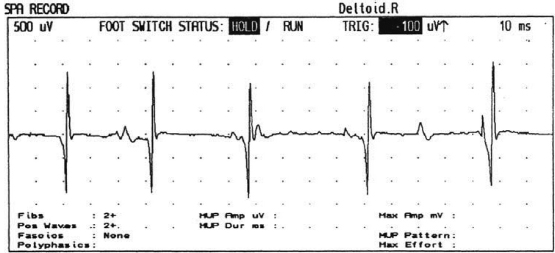
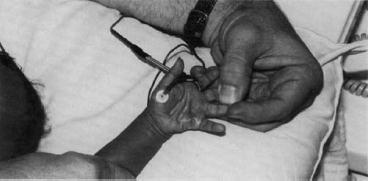
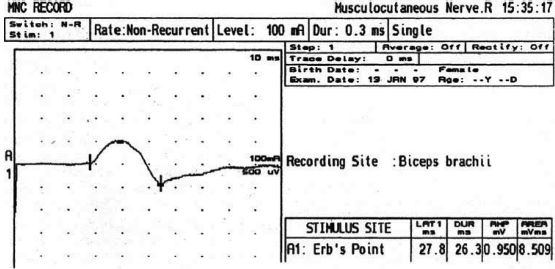
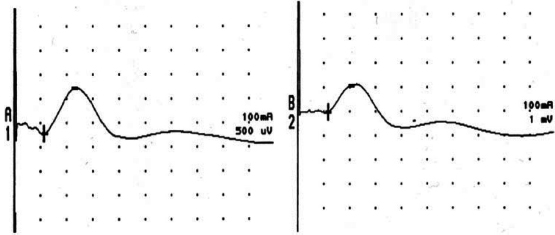
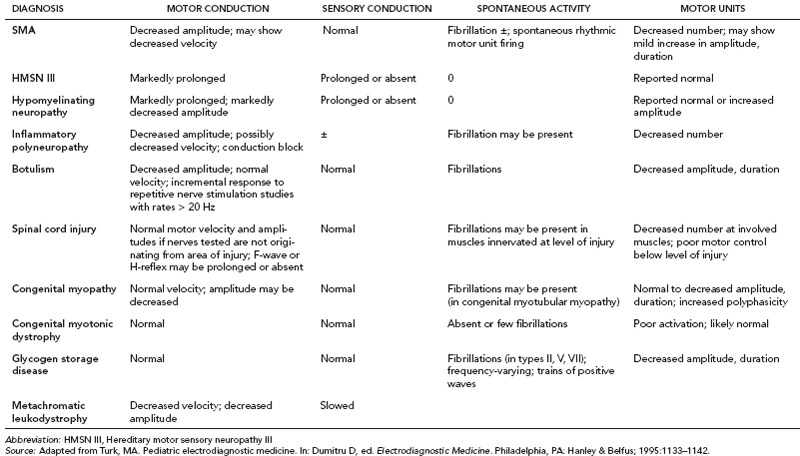
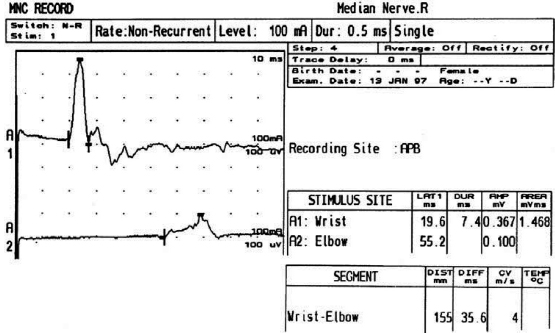
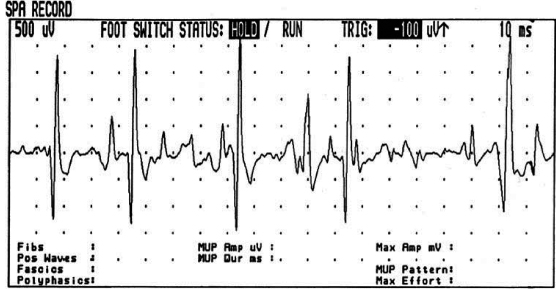
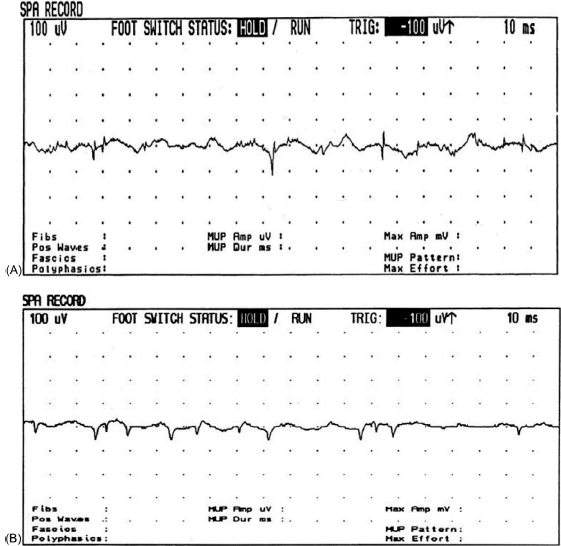
6 ELECTRODIAGNOSIS IN PEDIATRICS Craig M. McDonald INTRODUCTION Electromyography (EMG), nerve conduction studies (NCSs), and evoked potentials, including somatosensory evoked potentials (SSEPs) and motor evoked potentials (MEPs), provide useful information to assist the clinician in the localization of pathology within the lower motor neuron and selected areas of the central nervous system. In the case of acquired or hereditary disorders of the lower motor neuron—anterior horn cell, peripheral nerve, neuromuscular junction (NMJ; presynaptic or postsynaptic region), or muscle—electrodiagnostic studies (EDSs) are a useful tool to be used as an extension of the clinician’s physical examination. The information gained from EDSs may be invaluable in planning subsequent more invasive diagnostic studies (eg, muscle and nerve biopsy, cerebrospinal fluid (CSF) examination, MRI, which at times requires general anesthesia), allow for more cost-effective and specific molecular genetic testing, or aid in the surgical management of peripheral nerve trauma, compressive lesions, or entrapments. In the case of immune-mediated disorders such as myasthenia gravis or Guillain–Barré syndrome (GBS), EDSs may permit prompt treatment. While many may assume a decreased role of EDSs in the era of molecular genetic diagnosis for children with neuromuscular disorders, EDSs continue to play an important role in the diagnosis evaluation of children with neuromuscular disorders, although the practice paradigm is evolving. Major advances in genetic analysis and neuroimaging have modified the traditional diagnostic approach to neuromuscular disorders. A recent study investigated the role of pediatric EMG in the modern molecular era (1). All EMG studies performed at Boston Children’s Hospital from 2001 through 2011 were retrospectively reviewed and data were collected on study numbers, patient ages, referring provider sub-specialty, study indication, electrophysiologic diagnosis, and study utility. A total of 2,100 studies were performed. The volume increased from approximately 160 to about 250 studies/year over a 10-year period. There was a trend toward studying older children. Neurologists, including neuromuscular specialists, constituted the major referral pool, whereas referrals from orthopedics increased steadily. Polyneuropathies followed by mononeuropathies were the most common diagnoses referred to the electrodiagnostic laboratory. Fifty-seven percent of studies were normal. EMG provided meaningful information in 94% of cases. Another recent study assessed the diagnostic yield of electromyography in children with myopathic disorders (2), as the interpretation of pediatric electromyography in myopathic disorders is technically challenging. The investigators assessed the sensitivity and specificity of electromyography in pediatric myopathy cases through a retrospective chart review of patients 18 years and less, between 2009 and 2013. Two hundred twenty-four electromyographic studies were reviewed with the following referral diagnoses: myopathy, muscle weakness, neuromuscular disorders, myositis, myalgia, myoglobinuria, myasthenia, myotonia, cramps, periodic paralysis, hypotonia, and developmental delay. Only children who had an electromyography and muscle biopsy were included for analysis. Patients with neurogenic electromyography and NMJ disorders were excluded. Myopathic electromyography was defined as short-duration, low-amplitude, polyphasic motor unit potentials (MUPs) with rapid recruitment. Seventy-two patients were included (age range, 6 months–18 years) and they were grouped according to the following findings: group A: myopathic electromyography, and biopsy or genetically confirmed myopathy (32 cases); group B: myopathic electromyography but biopsy normal or nondiagnostic (12 cases); group C: normal electromyography but biopsy or genetically confirmed myopathy (three cases, all with metabolic myopathy); and group D: electromyography normal and biopsy normal or nondiagnostic (25 cases). The most common diagnoses were congenital myopathy (seven cases), metabolic myopathy (six cases), muscular dystrophy (six cases), genetically confirmed myopathy (five cases), myopathy, undefined (five cases), and inflammatory myopathy (four cases). The authors concluded that pediatric electromyography was 91% sensitive and 67% specific in myopathic disorders. The metabolic myopathies were commonly missed by electromyography. In another center, the sensitivity and specificity of preoperative electromyography were assessed for the detection of muscle pathology in using muscle biopsy findings as a gold standard. The sensitivity and specificity were determined to be only 58% and 56%, respectively (3). Clearly the diagnostic utility of pediatric EDSs is somewhat dependent on the skills of the electromyographer and the nature of the referral problems. Pediatric electrodiagnosis must be approached with knowledge of peripheral neuromuscular development and thoughtful planning of the study with regard to most likely diagnostic possibilities, developmental status of the child, and the likelihood that the pediatric electrodiagnostic practitioner will be able to provide clinicians and family with useful diagnostic information. The physical examination and developmental level of the infant or child direct the study. The examination requires patience and technical competence by an electrodiagnostic clinician experienced and skilled in the evaluation of children. This chapter will focus on considerations specific to the electrodiagnostic evaluation of infants and children with an emphasis on practical suggestions that may facilitate the completion of an accurate pediatric electrodiagnostic examination with the minimization of discomfort and distress to the child, parent, and pediatric electrodiagnostic specialist. MATURATIONAL FACTORS IN PEDIATRIC ELECTRODIAGNOSIS The normative neurophysiologic data relating to the maturation of peripheral nerves and muscle in children has greatly expanded in the recent past (4–13). The reader is referred to the volume by Jones, Bolton, and Harper (11) for an excellent review of neurophysiologic norms in pediatric populations. Peripheral nerve myelination begins at about the 15th week of gestation and continues throughout the first 3 to 5 years after birth (14). Conduction velocities (CVs) are determined by myelination, diameter of the fiber, and internodal differences. Myelination occurs at the same rate, whether intrauterine or extrauterine. CVs are directly related to gestational and postconceptual age and are unrelated to birth weight (15,16). CVs increase in direct proportion to the increase in diameter of fibers during growth. A direct relationship exists between the diameter of the axon and the thickness of the myelin sheath. The diameter of the fibers at the time of birth has been shown to be one-half of that in the adult. No unusual acceleration of myelination occurs subsequent to birth (17). Peripheral fibers reach their maximum diameter at 2 to 5 years after birth (17,18). The nodes of Ranvier continue to remodel with peak internodal distances being reached at 5 years of age. NERVE CONDUCTION STUDIES In general, normal standard adult values for CVs are reached by ages 3 to 5. In infancy, upper and lower extremity CVs are similar under age 1. Subsequently, faster conductions are maintained in the upper extremities, and comparatively slower conductions in the lower extremities as with adults. Unique values for expected CVs are observed for specific peripheral nerves. MOTOR NERVE CONDUCTION Motor conduction velocities (MCVs) in infants are found to be one-half of adult values. In infants, conduction studies should be at least greater than 20 M/S. At birth, mean MCVs for the median, ulnar, and peroneal nerves are 27 M/S. The median nerve may lag in maturation of CV relative to the ulnar and peroneal nerves. Ulnar MCV values reach the lower adult range by age 3 (17). The slight difference between ulnar and median MCV values present in the first 3 years of life disappears in children by 4 to 5 years of age. Careful and consistent measurements are necessary to achieve reliable and valid data. Normative values or selected motor nerve conduction velocities (NCVs) are shown in Table 6.1. DISTAL MOTOR LATENCY (DML) Distal motor latencies (DMLs) show maturational changes between infancy and 3 to 5 years of age, similar to MCVs. Normative data for distal latencies have generally been more incomplete with ranges of distances provided (from stimulation to active electrode). While the stimulation distance should always be recorded in the electrodiagnostic report, the specific distal latency is rarely of critical importance in determining a diagnosis in pediatric electrodiagnosis, as distal peripheral entrapments are relatively uncommon. Rather, reported distal latencies, which are either unusually fast or unusually slow in the setting of otherwise normal MCVs, should raise a suspicion regarding technical problems and identification of appropriate waveforms. The corrected DML may be used as an alternative for young children, by applying the formula of Slomic and colleagues found in Wagner and Buchthal (19): Corrected DML = measured DML − [L – X/MCV], where L = actual distance between stimulating cathode to the active recording electrode, and X = standard distance (4 cm for nerves of upper limbs and 5 cm for nerves of the lower limbs). Garcia and colleagues (13) have reported the most complete data to date on corrected DML in children (see Table 6.2). The corrected DML in the neonatal group is increased relative to other age groups, decreases over the first 12 months of life, remains unchanged between 12 months and 24 months, and slightly increases later. As most clinicians reading reports are not familiar with the corrected DML, an explanation of the calculation and normative interpretation should be included in the report, if this data is reported along with the actual DML and distance used. TABLE 6.1 NORMAL MOTOR CONDUCTION VELOCITIES (M/SEC) TABLE 6.2 CORRECTED DISTAL MOTOR LATENCY (MSEC) COMPOUND MUSCLE ACTION POTENTIAL (CMAP) Compound muscle action potential (CMAP) amplitudes are important to consider in the evaluation of axonal loss, conduction block, and muscle fiber atrophy. CMAP amplitudes of lower extremity nerves are one-half to one-third adult values in infants and upper extremity CMAPs may be one-third to one-fourth adult values during infancy. As with MCVs, CMAP amplitudes increase in size with age but adult values are generally not reached until the end of the first decade. Normal values for CMAP amplitudes are shown in Table 6.3. SENSORY NERVE CONDUCTION Modern EMG equipment, which includes amplifiers and signal averaging capability, allows sensory nerve action potentials (SNAPs) to be routinely recorded in the absence of peripheral nerve pathology. Maturational changes for orthodromic and antidromic sensory conduction are similar to that for motor fibers (9,20,21). In infants and young children, two distinct peaks are often observed in the SNAP with proximal stimulation. This two-peak potential has been attributed to differences in maturation between two groups of sensory fibers (19) and often persists until 4 to 6 years of age. Sensory NCVs may be calculated from single distal antidromic or orthodromic stimulations by measuring the distance from the stimulation point to the active electrode and the distal latency. Normative values for sensory NCVs in selected nerves using orthodromic stimulation and proximal recording are shown in Table 6.4. Normal values for orthodromic and antidromic SNAP amplitudes are shown in Table 6.5. F-WAVES The F-wave is a late response that appears as supermaximal motor nerve stimulation and arises from the discharge of a small number of motor neurons, in response to antidromic stimulation of the motor axon. The F-wave latency is measured from hand and foot intrinsic muscles and is useful for evaluating the motor NCV and proximal nerve segments. With the F-wave, the speed of motor nerve conduction is measured over a long distance, and therefore is less subject to errors inherent in the calculation of MCVs over short distances (10 cm or less). F-waves can be recorded from most limb nerves in newborns and young infants. The minimum F-latency in normal children recorded from hand muscles with median or ulnar nerve stimulation at the wrist is generally less than 20 msec in children younger than 6 years of age (9,10,22). In the lower extremities, the F-wave latency recorded from intrinsic foot muscles with peroneal or posterior tibial nerve stimulation at the ankle is generally less than 30 msec (7,9). Normal values for F-wave latencies for children are shown in Table 6.6. TABLE 6.3 NORMAL CMAP AMPLITUDES (MV) TABLE 6.4 NORMAL SENSORY CONDUCTION VELOCITIES (M/SEC) H-REFLEX The H-reflex is present in both the upper extremity (median and ulnar) and lower extremity (with posterior tibial stimulation) in infancy. While the tibial H-reflex persists into adulthood, the upper extremity H-reflex responses are present in virtually all infants at birth and become suppressed in most children over the course of the first year. Normal values for H-reflex latencies in children are shown in Table 6.7. NEUROMUSCULAR TRANSMISSION The NMJ shows less stability with repetitive nerve stimulation in normal newborns. At low rates of stimulation (1–2 Hz), no significant incremental or decremental changes in CMAP amplitude are observed (23). At higher rates of stimulation (5–10 Hz), normal infants may show slight facilitation. Decremental responses averaging 24% have been reported at high rates of stimulation (20 Hz) in normal newborn infants. At 50 Hz stimulation, normal newborns may show decrements of the order of 50% (20). In general, decremental changes greater than 10% at low rates of stimulation (2–5 Hz) and facilitatory changes greater than 23% at high rates of stimulation (20–50 Hz) are felt to be significant in the post-term infant (24). Some authors have utilized high rates of stimulation of the order of 50 Hz for 10 seconds to document facilitation greater than 20% to 23% (at times, over 100% increments are observed) in infantile botulism (24–26). TABLE 6.5 NORMAL SNAP AMPLITUDES IN CHILDREN (μV) ELECTROMYOGRAPHY MOTOR UNIT CONFIGURATION AND AMPLITUDE Amplitudes of motor unit action potentials (MUAPs) are lower in infants with amplitudes ranging from 150 microvolts to approximately 2,000 microvolts. Generally, MUAPs more than 1,000 microvolts in 0 to 3-year-old children are rare (27,28). In infants, MUAPs are usually biphasic or triphasic. MOTOR UNIT DURATION Infantile MUAPs are often shorter in duration. De Carmo (27) found newborn infants to exhibit durations 17% to 26% shorter than those seen in adults. Durations of MUAPs are often shorter than 5 msec in infants. MOTOR UNIT RECRUITMENT In very young infants and children, it is difficult to assess the strength of voluntary contraction and determine when the interference pattern is full. In general, as the strength of voluntary contraction increases, there is an increase in MUAPs recruited. However, the recruitment pattern in infants may be disordered and chaotic. As with adults, the recruitment frequency, defined as the firing rate of an MUAP when a different MUAP first appears with gradually increasing strength of voluntary contraction, is helpful in differentiating a myopathic process (lower recruitment frequency values) from a neuropathic process (higher recruitment frequencies greater than 20–25 Hz). An example of neuropathic recruitment is shown in Figure 6.1. TABLE 6.6 NORMAL F-WAVE LATENCIES IN CHILDREN (MSEC) TECHNICAL FACTORS WITH INFANTILE NERVE CONDUCTION STUDIES TEMPERATURE The maintenance of appropriate subject temperature is essential during NCSs. Neonates generally have difficulty with temperature homeostasis and low subject temperature may have profound effects on CVs. A skin temperature of 36 to 37°C produces near-nerve temperatures of 37 to 38°C and avoids spurious reductions in NCVs and prolongation of distal latencies. It is assumed that a 1°C drop in temperature produces a slowing of conduction of the order of 2 to 3 m/s. Every attempt should be made to maintain extremity temperature with infant warmers, heating lamps, or warm blankets. VOLUME CONDUCTION Volume conduction is defined as the current transmission from a potential source through a conducting medium, such as the body tissues. This may produce depolarization of peripheral nerves in proximity to the specific nerve being studied, and this is particularly problematic in smaller children with less soft tissue separating nerves. For example, volume conduction can produce simultaneous stimulation of both the median and ulnar nerves at the wrist or at the elbow. Such volume conduction should always be suspected when higher stimulation intensities or durations are utilized and when CMAP configurations show an initial positive deflection or a multiple peak configuration. TABLE 6.7 NORMAL H-REFLEX LATENCIES IN CHILDREN (MSEC) SHOCK ARTIFACT Shock artifact is a common problem with smaller subjects because of short distances between the stimulator and recording electrodes. This may be particularly problematic with distal stimulation. The ground electrode should be placed between the stimulating and recording electrodes and, in infants, often a standard 6 mm silver disk or ring electrode can be placed around the wrist or ankle. Alternatively, the ground disk may be taped to the dorsal surface of the hand. Other approaches to minimize shock artifact in young children include the utilization of pumice paste to reduce skin impedance and permit suprathreshold stimulation with lower electrical currents, use of a minimal amount of conduction gel or cream, and rotation of the proximal anode in relation to the distal cathode. FIGURE 6.1 Neuropathic recruitment of the deltoid in a 12-month-old child with a brachial plexus injury sustained at birth. The initial recruited motor unit action potential is 2,500 µV, and it is firing at 25 Hz. MEASUREMENT OF DISTANCES/MEASUREMENT ERROR Distance measurements must be extremely meticulous during pediatric electrodiagnostic evaluations. Segment studies are often of the order of 6 to 10 cm in length. A measurement discrepancy of only 1 cm may produce as much as a 10% to 15% conduction velocity error. STIMULATING ELECTRODES For neonates and young infants, small stimulators with short interelectrode distances are commercially available and simplify the testing of short nerve segments over small extremities (Figure 6.2). The stimulation intensity may be reduced by the use of a small monopolar needle electrode as the stimulating cathode with a more proximal surface anode in close proximity. For example, for ulnar orthodromic sensory studies, the author has utilized ring electrodes on the fifth digit and recording electrodes over the ulnar nerve at the elbow. Generally, a standard bipolar stimulator may be utilized for children 6 months of age and older. FIGURE 6.2 Pediatric nerve stimulator (A). The interelectrode distance between cathode and anode is less than 2 centimeters (B). RECORDING ELECTRODES Sensory Conduction Generally, SNAPs are easily recorded in newborns. The standard ring electrodes, needle recording electrodes, and/or pediatric-size finger-clip electrodes may be used. While for adults, a 4 cm interelectrode distance is optimal, this is not possible in small children. Hence, the pediatric electrodiagnostic clinician should attempt to obtain as much distance as possible between active and reference electrodes. Every attempt should be made to obtain at least a 2 cm interelectrode distance. Stimulation of the digits, palm, or wrist with electrodes located more proximally at the elbow for median and ulnar sensory studies provides longer distance and less measurement error. In general, normative data for sensory NCVs are more readily available than normative data for distal latencies at specific distances. Motor Conduction Generally, standard 6 mm silver disk surface electrodes are used as active and reference electrodes for motor conduction studies. Some electrode diagnosticians prefer the use of ring electrodes on digits as the reference electrode and a standard surface electrode over the Moro point at the muscle as the active electrode (see Figure 6.3). Often 4 to 6 cm distances are used from the stimulator to the active electrode. CVs and CMAP amplitudes are generally more relevant data in infants than motor distal latencies because distal nerve entrapments are rare. Thus, the distances used from distal stimulation to the active electrode are less critical. FIGURE 6.3 Recording electrodes for a median motor nerve conduction study in a small child. The active electrode is placed over the abductor pollicis brevis on the thenar eminence. The recording electrode is a ring electrode placed on the index finger. The ground electrode is a 6-millimeter silver disc electrode placed on the back of the hand. SPECIAL CONSIDERATIONS FOR NERVE CONDUCTION STUDIES The best normative data for pediatric NCSs are available for the median, ulnar, peroneal, tibial, facial, and phrenic motor nerves and the median, ulnar, and sural sensory nerves. Stimulation of the posterior tibial nerve (recording abductor hallucis brevis) produces a discrete CMAP more commonly than stimulating the peroneal nerve (recording over extensor digitorum brevis). The EDB muscle may be difficult to visualize or palpate in infants. Its CMAP configuration frequently has either an initial positivity or a low broad configuration. In addition, the CMAP amplitude may change substantially with slight changes in position for the active electrode over the extensor digitorum brevis. The axillary and musculocutaneous motor NCSs may be helpful in the setting of infantile brachial plexopathy. Care should be taken to minimize volume conduction. Often, the intact side is used for amplitude comparisons. Evaluations of proximal nerves such as the axillary spinal accessory musculocutaneous and femoral are often useful in the evaluation of severe demyelinating neuropathies (see Figure 6.4). The distal latencies of these nerves may be severely prolonged on the setting of severe reductions in the CMAPs of more distal nerves due to conduction block or axon loss. Percutaneous stimulation of the phrenic nerve is performed with techniques similar to those utilized in the adult, with stimulation performed at the posterior border of the sternocleidomastoid at the level of the thyroid cartilage or alternatively just medial (or occasionally lateral) to the sternal head of the sternocleidomastoid. Recording electrodes may be placed in the fifth to sixth intercostal space 2 cm apart at the anterior axillary line or alternatively an active electrode may be placed immediately below the costal margin at the level of the nipple with the recording electrode at the xiphoid. The active electrode may need to be moved to adjacent positions to obtain an optimal M-wave (see Figure 6.5). Normative values for phrenic latencies have been reported in children (29,30). The author prefers to use ultrasound visualization of the diaphragm simultaneously with phrenic nerve stimulation to confirm downward deflection of the diaphragm. Volume conduction to the long thoracic nerve may produce a CMAP from the serratus anterior rather than the diaphragm. The downward deflection of the diaphragm spontaneously and with electrical stimulation may be confirmed, and the distance of diaphragmatic excursion quantitatively measured by ultrasound M-mode (31). REPETITIVE NERVE STIMULATION STUDIES Every attempt should be made to stabilize the extremity with an infant or pediatric-size arm board. The author prefers to use a block electrode or surface cathode and anode electrodes taped over the nerve as opposed to a handheld stimulator. This helps standardize each stimulation during a train of five stimuli at low or high rates of stimulation. In newborns, the author prefers to stimulate the median or ulnar nerve at the elbow to minimize shock artifact. Care should be taken to obtain a stable baseline between stimulations in a train. Decrements or increments in amplitude should be accompanied by similar decrements or increments in the area. If no concomitant area changes occur, then technical factors (changing baseline or changing temporal dispersion) may explain a decrement or increment in amplitude. FIGURE 6.4 Nerve conduction study of the musculocutaneous nerve in Charcot-Marie-Tooth (CMT) type III. The nerve is stimulated at Erb’s point and the recording electrode is placed over the biceps brachii. Distal latency is severely prolonged at 27.8 milliseconds. Note the reduced compound muscle action potential amplitude, presumably due to conduction block, and the relative lack of temporal dispersion, which is frequently seen in CMT. FIGURE 6.5 Phrenic nerve conduction study in a 13-year-old child with C2 traumatic spinal cord injury. A1 is the compound muscle action potential (CMAP) amplitude obtained on the right side and B2 is the CMAP obtained on the left side. Latencies are approximately 5 milliseconds and amplitudes from baseline to peak 1 mV. The viability of the phrenic nerves allowed placement of a phrenic nerve–diaphragm pacer for ventilation. TECHNICAL FACTORS OF NEEDLE ELECTROMYOGRAPHY ELECTRODES Generally, 26 to 28 gauge Teflon-coated monopolar electrodes, usually 25 mm in length, are utilized. Some laboratories routinely use disposable concentric facial needle electrodes. These electrodes have smaller calibrated recording areas and hence provide more stability of MUAP configuration. In addition, concentric needle electrodes are more sensitive to changes in duration and amplitude than monopolar needle electrodes. Use of smaller electrodes (either small monopolar needles or small diameter concentric needle electrodes originally designed for the examination of adult facial muscles) provides considerable psychological advantages in children of sufficient developmental age to associate needles with pain. The instrumentation utilized for needle EMG of children is essentially the same as that used in adults. In the Intensive Care Unit, electrical interference may necessitate the use of either a facial concentric needle or a needle reference electrode. Long electrodes or long electrode leads can create problems with ambient electrical interference. OPTIMAL MUSCLES TO STUDY FOR REST ACTIVITY In evaluating an infant or young child for a generalized disorder, specific muscles are chosen to permit evaluation of insertional and spontaneous activity. The distal hand (first dorsal interosseous) and foot muscles of infants usually have minimal voluntary activity due to immature motor control at this developmental age, making them good sites to assess spontaneous activity. In addition, extensor muscles such as the vastus lateralis and gastrocnemius in the legs and the triceps in the upper extremities are useful sites for the evaluation of insertional and spontaneous activity. In the neonate and young infant, foot and hand intrinsic muscles exhibit high levels of endplate noise because of the relatively larger endplate area in the immature muscle. This endplate activity may be confused with fibrillation potentials. Fibrillation potentials and positive sharp waves are not typically observed in the full-term normal newborn. OPTIMAL MUSCLES FOR EVALUATION OF RECRUITMENT, MOTOR UNIT CONFIGURATION, AND INTERFERENCE PATTERN In general, flexor muscles such as the tibialis anterior and the iliopsoas are useful for the evaluation of MUAPs and recruitment in the lower extremity. These muscles can be activated by tickling or pinching the bottom of the foot, producing a withdrawal response. In the upper extremity, the flexor digitorum sublimis and biceps muscles are frequently reflexively activated by the newborn or young infant. More proximal muscles can be activated by moving the extremity or positioning the extremity to produce antigravity stabilization of the limb by the firing of proximal musculature. Alternatively, reflex posturing techniques such as the Moro response can be used to activate the shoulder abductors but are usually not necessary. SEDATION Pediatric physiatrists and neurologists performing pediatric electrodiagnostic evaluations have noted that extreme behavioral distress most frequently occurs among 2 to 6 year olds (32,33). Pain medications are occasionally or always prescribed by 50% of pediatric electromyographers (33). General anesthesia is occasionally utilized by 25% of electrodiagnostic practitioners (33). One study demonstrated that children exhibiting more behavioral distress during pediatric electrodiagnostic evaluations were younger, had been uncooperative with previous painful procedures, were more likely to have had more negative medical/dental experiences, and had mothers who themselves reported greater fear and anxiety about undergoing EMG/NCSs (32). While some electromyographers never utilize sedation, there has been more interest in the use of analgesia, conscious and deep sedation, and more recently, general anesthesia with propofol or inhalational anesthetics. Traditional sedative choices include chloral hydrate (50–100 mg/kg), “DPT” (Demerol [meperidine hydrochloride], Phenergan [promethazine], and Thorazine [chlorpromazine]) and midazolam hydrochloride nasal spray. EMLA cream (lidocaine 2.5% and prilocaine 2.5%) has been used during electromyographic evaluations as a topical anesthetic (34). The mean duration of topical application in infants or older children was 45 to 145 minutes. Greater pain relief was obtained with the use of EMLA over the extensor forearm than the thenar eminence. While general anesthesia is usually not necessary, the author has increasingly involved critical care and anesthesia colleagues who have utilized either propofol (2,6-diisopropylphenol), an intravenous sedative–hypnotic agent, or inhalational anesthetics with laryngeal mask anesthesia (LMA) airways for the electrodiagnostic evaluation of 18-month-old infants to 6-year-old children who exhibit substantial behavioral distress during an initial attempt at an electrodiagnostic evaluation without sedation. Propofol produces rapid onset of anesthesia (in 1–3 minutes) and sedation is maintained by either a continuous infusion or multiple boluses. Subjects usually awaken in less than 10 minutes from the time the infusion is discontinued. Sedation, analgesia, and particularly, general anesthesia have inherent risks and require appropriate monitoring. Propofol should be administered by an anesthesiologist or pediatric intensivist prepared to bag-mask ventilate or intubate the child if necessary. Adequate monitoring generally requires a sedation suite, pediatric ICU, and a recovery room or operating room. The author typically obtains all NCSs and a thorough examination of multiple muscle sites for abnormal spontaneous rest activity while the subject is deeply sedated or anesthetized with propofol. The level of sedation is then titrated to a point where appendicular movement is elicited with needle insertion or stimulation of the extremity. At this point, under lighter sedation, recruitment pattern and motor unit configuration are assessed. As the child awakens, interference pattern is evaluated with more vigorous motor activity. Children are usually amnestic to the EMG examination subsequent to propofol anesthesia. The cost of anesthesia must be weighed against the importance of the acquisition of a thorough, technically precise, and accurate electrodiagnostic evaluation. An EMG obtained under anesthesia usually provides a suboptimal evaluation of motor unit configuration, recruitment pattern, and interference pattern with maximal effort, but better evaluation of quiet muscle for spontaneous activity and a more comprehensive acquisition of NCSs and repetitive nerve stimulation studies. The key to successful data acquisition in most pediatric electrodiagnostic evaluations remains a well-organized, well-planned approach with distinct diagnostic questions prospectively considered. If the examination is planned to answer a specific question, it is usually possible to proceed expeditiously, completing the examination within a reasonable time (30 minutes). As children approach 6 years of age, it becomes easier to talk them through an evaluation and elicit their participation and cooperation. NCSs are usually better tolerated than needle electromyography and many pediatric electromyographers perform NCSs first. Increased behavioral distress, subsequent to a needle examination, makes the motor nerve conductions and particularly, sensory NCSs, technically difficult due to excessive EMG background noise. LIMITATIONS OF SINGLE-FIBER EMG While normative data for fiber density, mean consecutive difference, and jitter have been reported for different muscles among different pediatric age groups (35), this procedure is difficult to use in younger children with limited ability to cooperate. Alternatively, a stimulated single-fiber EMG study may be obtained under general anesthesia in those suspected of a congenital myasthenic syndrome, and this technique has yielded excellent sensitivity and specificity for the identification of a neuromuscular transmission disorder (36–38). SPECIFIC CLINICAL PROBLEMS IN PEDIATRIC ELECTRODIAGNOSIS ELECTRODIAGNOSTIC EVALUATION OF THE FLOPPY INFANT The most common referral for an electrodiagnostic examination in the infant is generalized hypotonia. The most common etiology for infantile hypotonia is central, accounting for approximately 80% of cases. A differential diagnosis of infantile hypotonia is shown in Table 6.8 (39). Electrodiagnostic abnormalities in selected conditions producing infantile hypotonia are shown in Table 6.9. Neurogenic causes of generalized weakness in infants are more accurately diagnosed with EDSs than are myogenic causes (40–42). A study of the predicted value of the electrodiagnostic examination in the hypotonic infant showed that EDSs accurately predicted the diagnosis in 65% of infants with spinal muscular atrophy (SMA) and only 10% of infants with myopathy. Seventy-five percent of the EDSs performed on infants with documented myopathies were considered normal (43). The sensitivity of EMG improves after age 2 (42). TABLE 6.8 DIFFERENTIAL DIAGNOSIS OF INFANTILE HYPOTONIA TABLE 6.9 INFANT HYPOTONIA: ELECTRODIAGNOSTIC ABNORMALITIES In the evaluation of hypotonia, a complete electrodiagnostic evaluation is useful, including motor and sensory NCSs and appropriate needle examination with the highest yield muscles examined initially, and, if necessary, repetitive nerve stimulation. It should be emphasized that NCSs and electromyography are an extension of the clinician’s physical examination. Electrodiagnostic findings need to be interpreted in light of clinical examination findings. Care should be taken not to overinterpret subtle findings on needle electromyography. Low-amplitude, short-duration, polyphasic MUAPs, which would be considered myopathic in adults, may be normal in young children. Motor unit amplitudes and durations may be reduced in the normal young child and mistaken for myopathic MUAPs. Endplate noise, abundant in the small intrinsic muscles of the hand and foot may be difficult to distinguish from fibrillation potentials. Thus, borderline findings on needle EMG should not be overinterpreted in the infant and young child. Parents should be cautioned prior to an electrodiagnostic evaluation that definitive diagnostic information is often not obtained, and the results may help guide further diagnostic studies. For example, results from EMG may help to guide further studies such as muscle biopsy by providing information about the most appropriate muscle site for the biopsy. With SMA, an electrodiagnostic evaluation can allow the clinician to defer a muscle biopsy and proceed with molecular genetic studies of the survival motor neuron (SMN) gene. Often the SMN gene is ordered prior to any EDSs being performed, so fewer studies have been performed on this population over the past decade. EDSs in patients with hereditary motor–sensory neuropathy help to categorize the neuropathy as either primarily demyelinating or axonal, and such information may help focus subsequent molecular genetic analyses. In general, nerve conduction and electromyography still provide a useful tool for the localization of lesions within the lower motor neuron, but fewer studies have been required as genetic studies have become commercially available. DIFFERENTIAL DIAGNOSIS FOR EARLY RESPIRATORY DISTRESS IN INFANCY The differential diagnosis of lower motor neuron disorders with perinatal respiratory distress is fairly limited. Generally, respiratory distress within the first few days of life can be seen in SMA type I, congenital hypomyelinating neuropathy, congenital myasthenia, transient neonatal myasthenia, congenital myotonic muscular dystrophy, neurogenic arthrogryposis, and x-linked myotubular myopathy. These disorders are easily differentiated with EDSs and in some instances molecular genetic findings. For example, congenital myotonic muscular dystrophy may be definitively diagnosed with molecular genetic studies at the chromosome 19q13.3 locus. In congenital hypomyelinating neuropathy, sensory conduction abnormalities are unrecordable and motor NCVs are markedly slowed down (2–5 m/s) with temporal dispersion and low-amplitude evoked potentials (see Figure 6.6). SMA patients show normal sensory conductions, decreased CMAP amplitudes, occasional fibrillations, and decreased numbness of MUAPs. Congenital myasthenia patients show normal sensory conductions, normal motor NCVs, and abnormalities on repetitive nerve stimulation studies. X-linked myotubular myopathy patients show profuse fibrillations and myopathic MUAPs on EMG and diagnosis is confirmed by muscle biopsy. ACUTE ONSET INFANTILE HYPOTONIA Acute onset hypotonia in a previously normal infant should warrant an evaluation to rule out acute inflammatory demyelinating polyneuropathy (AIDP), infantile botulism, infantile polymyositis, an infantile form of myasthenia, a toxic process, or acute onset myelopathy. Repetitive motor nerve stimulation studies should be performed under the following circumstances: (a) there is constipation, bulbar involvement, and/or respiratory distress; (b) an infant presents with ptosis or extraocular muscle weakness; (c) CMAP amplitudes are severely reduced; (d) “myopathic” MUAPs are present; (e) a repetitive CMAP is observed after single supramaximal stimulation on routine NCS, suggestive of a diagnosis of congenital myasthenia with congenital acetylcholinesterase (AChE) deficiency or classic slow-channel syndrome. MOTOR NEURON DISORDERS (EG, SMA) SMA is perhaps the most common lower motor neuron disorder causing infantile hypotonia. The predictive value of needle electromyography (EMG) in the diagnosis of SMA has been established (40–43), but the need for EDSs has diminished over the years, given the 95% or greater sensitivity of SMN gene studies. As SMA remains an important consideration in infantile hypotonia, a review of the electrodiagnostic findings is useful. The findings in this motor neuron disorder have largely been consistent with motor axonal loss, denervation, and (among persons less severely affected) reinnervation. Traditional electrodiagnostic criteria for motor neuron disease are not suitable for patients with childhood SMA. For example, Buchthal (45) found that many infants with SMA did not meet strict criteria for motor neuron disease. If clinical findings suggest SMA, study of at least two muscles innervated by different nerve roots and peripheral nerves in at least three extremities is indicated (46). In the infant, spontaneous activity may be more readily determined with the study of muscles that are not as commonly recruited, such as the vastus lateralis, gastrocnemius, triceps, and first dorsal interosseous. Recruitment and motor unit characteristics can be assessed in muscles that are readily activated such as the anterior tibialis, iliopsoas, biceps, and flexor digitorum sublimis (45). The paraspinal muscles are usually not studied due to poor relaxation, and the experienced pediatric electrodiagnostic medicine consultant usually defers needle evaluation of the tongue in the hypotonic infant. FIGURE 6.6 Median nerve conduction in a 5-year-old child with congenital hypomyelinating neuropathy documented by sural nerve biopsy and molecular genetic studies of the EGRF 2 gene. Distal latency is markedly prolonged at 19.6 milliseconds. There is reduced compound muscle action potential amplitude, at 0.367 mV, conduction block (note the drop in amplitude from distal to proximal), and conduction velocity at 4 m/s. Although some authors (46) have described high-density fibrillation potentials in infants with poorer outlook, most studies have not demonstrated abundant fibrillation potentials in the infantile form (46–48). In SMA III, the incidence of fibrillation potentials ranged from 20% to 40% in one series (49) to 64% in another (50). The incidence of fibrillation potentials in SMA III does not approach the level seen in SMA 1. Additionally, spontaneous activity has been more frequently observed in the lower extremities than upper limbs, and proximal more than distal muscles in SMA III (49). The degree of spontaneous activity has not been found to be independently associated with a worse prognosis in SMA (43). Fasciculations are uncommonly observed in SMA Type I and appear more commonly in SMA II and III (46–48). In younger patients, fasciculations are difficult to distinguish from spontaneously firing MUAPs. In relaxed muscles, some motor units exhibit a spontaneous rhythmic firing (47,48,51). Voluntary MUAPs frequently fire with an increased frequency although recruitment frequency may be difficult to determine consistently in infants. Compared to age-matched norms, MUAPs show longer duration, particularly in older subjects, and higher amplitude; however, a bimodal distribution may be seen with some concomitant low-amplitude short-duration potentials (47). Large-amplitude, long-duration MUAPs may be absent in many infants with SMA I but more commonly observed in SMA II and III (46). The percentage of large-amplitude MUAPs increase with the duration of the disease (48). Other signs of reinnervation, such as polyphasic MUAPs may be observed in more chronic and mild SMA. These polyphasic MUAPs may include late components such as satellite or nascent potentials. There may also be temporal instability of the waveform observed in individual MUAPs. Reduced recruitment (an incomplete interference pattern) with maximal effort is perhaps the most consistent finding in all SMA types (see Figure 6.7). In one series (43), the amplitude of MUAPs and degree of decrement in recruitment pattern were not individually associated with worse prognosis. Motor NCVs and CMAP amplitude have been shown to be reduced in many patients with infantile SMA. The degree of motor conduction slowing (if present) tends to be mild and greater than 70% of the lower limit of normal (48,50,52–54). Reduction of motor conductions to less than 70% of the lower limit of normal is described as an exclusionary criterion for SMA (55). The mild slowing of motor conductions is present to the same degree over distal and proximal segments as determined by M- and F-wave responses (53). The slowing of conduction is generally seen in those with correspondingly low-amplitude CMAPs and is thought to be due to selective loss of the fastest conducting fibers from large motor units. Alternatively, arrested myelination in utero has been proposed to explain this slowing in motor conduction noted in some SMA cases at birth (43). Survival has been found to be longer for those SMA infants with normal MCVs over a distal segment (43). Significant reductions in CMAP amplitudes have been frequently reported for SMAs I to III (43,46,50). Kuntz (50) reported a tendency toward greater reductions in CMAP amplitude among patients with earlier age of onset and shorter survival. FIGURE 6.7 Incomplete or reduced interference pattern in spinal muscular atrophy type II. Note the large amplitude motor unit action potential (3,000 µV) firing at 25 Hz. Sensory NCSs in SMA show essentially normal sensory CVs and SNAP amplitudes. Significant abnormalities in sensory studies exclude a diagnosis of SMA (55), while minor abnormalities in sensory CVs have infrequently been noted in SMA (52,56,57). Such rare sensory abnormalities have not been reported in SMA patients with diagnostic confirmation by molecular genetic studies. CMAP amplitude and motor unit number estimation (MUNE) have increasingly been used in recent years as potential biomarkers for clinical trials in SMA. In one study (58), denervation was assessed in 89 SMA I, II, and III subjects via MUNE and maximum compound muscle action potential (CMAP) studies, and results correlated with SMN2 copy, age, and function. MUNE and maximum CMAP values of the ulnar nerve were distinct among SMA subtypes. Changes in MUNE and maximum CMAP values over time were dependent on age, SMA type, and SMN2 copy number. SMN2 copy number less than 3 (consistent with SMA I) was correlated with lower MUNE and maximum CMAP values and worse functional outcomes. As SMN2 copy number increased, so did functional status. Change in MUNE longitudinally over the time intervals examined in this study was not statistically significant for any SMA cohort. However, a decline in maximum CMAP over time was apparent in SMA2 subjects. Age-dependent decline in MUNE and maximum CMAP was apparent in both SMA I and SMA II subjects, with age as an independent factor regardless of type. Maximum CMAP at the time of the initial assessment was most predictive of functional outcome. Prospective longitudinal studies in four prenatally diagnosed infants demonstrated significant progressive denervation in association with symptomatic onset or functional decline. In another study (59), MUNE values correlated with Hammersmith Functional Motor Scale (HFMS) scores. Increased single motor unit action potential (SMUP) amplitude values correlated with decreased HFMS scores. The study confirmed that the MUNE method is a useful tool reflecting motor unit loss in SMA, and it is easy to perform and well tolerated. In a recent longitudinal study, 62 children with SMA types II and III were observed prospectively for up to 42 months (60). Longitudinal electrophysiologic data were collected, including compound motor action potential (CMAP), SMUP amplitude, and MUNE. Significant motor neuron loss and compensatory collateral reinnervation were noted at baseline. Over time, there was a significant mean increase in MUNE (4.92 units/year), a mean decrease in SMUP amplitude (−6.32 μV/year), and stable CMAP amplitude. The unexpected longitudinal results differed from findings in amyotrophic lateral sclerosis studies, perhaps indicating that compensatory processes in SMA involve new motor unit development. A better understanding of the mechanisms of motor unit decline and compensation in SMA will be important for assessing novel therapeutic strategies and for providing key insights into disease pathophysiology. MUNE and CMAP amplitudes seem to be sensitive parameters reflecting motor dysfunction in SMA but additional longitudinal studies are needed. To investigate these measures as biomarkers of treatment response, MUNE and sciatic CMAP measurements were obtained in SMNΔ7 mice treated with antisense oligonucleotide (ASO) or gene therapy (61). ASO-treated SMNΔ7 mice were similar to controls at days 12 and 30. CMAP reduction persisted in ASO-treated SMNΔ7 mice at day 12 but was corrected at day 30. Similarly, CMAP and MUNE responses were corrected with gene therapy to restore SMN. SMN restoring therapies result in preserved MUNE and gradual repair of CMAP responses. This provides preclinical evidence for the utilization of CMAP and MUNE as biomarkers in future SMA clinical trials. SPINAL CORD INJURY Neonatal spinal cord injury (SCI) may occur as an obstetrical complication or as a result of a vascular insult to the spinal cord. Typical clinical presentation may include findings of diffuse hypotonia, possible respiratory distress, hyporeflexia, and urinary retention. Bilateral flaccid paralysis of the upper extremities may be a sign of neonatal SCI (62). An anterolateral SCI due to a vascular insult will produce EMG findings of severe denervation in diffuse myotomes. Typically, 2 to 3 weeks may lapse before fibrillations and positive sharp waves are elicited. Anterior horn cell and axonal degeneration will typically result in decreased CMAP amplitudes in multiple peripheral nerves. SNAP amplitudes are spared. Somatosensory evoked potentials may be spared if posterior columns are preserved. Traumatic SCI often results in loss of anterior horn cells at a specific “zone of injury.” For example, a child with C5 tetraplegia may have denervation present at the bilateral C6 and C7 myotomes. This zone of partial or complete denervation becomes particularly relevant in the evaluation of a patient for possible placement of an implanted functional electrical stimulation system for provision of voluntary grasp and release. The presence of denervation necessitates concomitant tendon transfers with electrical stimulation of the transferred muscle group. Somatosensory evoked potentials (SSEPs) may help establish a sensory level in an infant or young child with SCI and is also useful in the evaluation of the comatose or obtunded child at risk for SCI without radiographic abnormality (SCIWORA). Somatosensory evoked potentials are discussed in the following. Transcranial electrical MEPs to monitor the corticospinal motor tracts directly are now used routinely in addition to SSEPs for detection of emerging SCI during surgery to correct spine deformity or resect intramedullary tumors (63–67). Afferent neurophysiologic signals can provide only indirect evidence of injury to the motor tracts since they monitor posterior column function. Transcranial electrical MEPs are exquisitely sensitive to altered spinal cord blood flow due to either hypotension or a vascular insult. Moreover, changes in transcranial electrical MEPs are detected earlier than are changes in SSEPs, thereby facilitating more rapid identification of impending SCI. BRACHIAL PLEXUS AND CERVICAL NERVE ROOT LESIONS Traumatic obstetrical brachial plexopathy or “neonatal brachial plexus palsy” (NBPP) usually results from traction on the brachial plexus (predominantly upper trunk) and its associated spinal roots. This can lead to stretching or rupture of the trunks of the plexus and/or partial axonotmesis or avulsion of the spinal roots. The most common cause is a shoulder dystocia of the anteriorly presenting shoulder causing excessive lateral neck traction. Injury to the upper trunk of the brachial plexus, and/or C5 and C6 cervical roots is the more common injury known as Erb–Duchenne palsy. Damage to the lower trunk, and/or C8-T1 cervical roots, is referred to as Klumpke’s palsy. Severe brachial plexus injuries may involve the entire plexus and C5-T1 nerve roots diffusely. Horner’s syndrome due to injury of the C8 and T1 roots and the superior cervical sympathetic ganglion may be an associated clinical finding. An isolated Klumpke’s palsy is rare in the setting of traumatic birth palsy and usually results from a fall onto a hyperabducted shoulder, penetrating trauma, or tumor. EDSs help determine the location (root and/or plexus), extent, and severity of the brachial plexus injury. Examination should be deferred until at least 3 to 4 weeks after the injury to allow for abnormal spontaneous rest activity (fibrillations and positive sharp waves) to develop in the setting of denervation and axon loss (see Figure 6.8). Complete injuries are characterized electromyographically by absent MUAPs and absent CMAP amplitudes in peripheral nerves supplied by the transected axons. In the setting of total motor paralysis, motor NCSs with measurement of the amplitude of the CMAPs in distal and proximal muscles provide useful prognostic information. For example, the preservation of the CMAP amplitude 10 days or more after the injury with complete clinical paralysis suggests that the damage is, in part, a neuropraxic injury with better prognosis. In this setting, F-waves are absent. If motor function is absent and no MUAPs are observed, examination of the amplitude of the SNAPs in the dermatomal distribution of the branches of the affected brachial plexus trunks can help distinguish injuries to the plexus from severe cervical root injuries or avulsions. The sensory dorsal root ganglion lies in the intervertebral foramen distal to the damaged segment with a root injury, leaving the sensory axon projection from the dorsal root ganglion to the limb intact. Thus, the SNAP is obtainable in the setting of a root avulsion with absent clinical sensation.





Related posts:
 Psychological Assessment in Pediatric Rehabilitation
Acute Management and Rehabilitation of Sport-Specific Musculoskeletal Injuries
Neurodegenerative and Demyelinating Diseases and Other CNS Disorders
Neuromuscular Diseases
Language Development and Disorders of Communication and Oral Motor Function
Orthotics and Assistive Devices
Psychological Assessment in Pediatric Rehabilitation
Acute Management and Rehabilitation of Sport-Specific Musculoskeletal Injuries
Neurodegenerative and Demyelinating Diseases and Other CNS Disorders
Neuromuscular Diseases
Language Development and Disorders of Communication and Oral Motor Function
Orthotics and Assistive Devices
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


