Cystic Fibrosis
Beryl J. Rosenstein
Cystic fibrosis (CF) is the most common life-shortening genetic disease affecting populations of European origin. The triad of chronic obstructive pulmonary disease, pancreatic exocrine deficiency, and abnormally high sweat electrolyte concentrations is present in most patients. CF is the major cause of chronic debilitating pulmonary disease and pancreatic exocrine deficiency in patients in the first three decades of life and accounts for a significant number of cases of neonatal intestinal obstruction. The name of the disease is derived from the characteristic histologic changes seen in the pancreas.
GENETICS
Estimates of the incidence of CF vary according to the population studied, but a reasonable figure for whites is 1 in 3,300 live births. The incidence in blacks in the United States is 1 in 16,300; in Asian Americans, it is 1 in 32,100. Transmission is autosomal recessive. On the basis of incidence figures, 1 in 29 whites in the United States is estimated to be a carrier (heterozygote) of a CF mutation. A heterozygote advantage has been postulated but never documented.
The gene responsible for CF spans 250,000 base pairs of genomic DNA located on the long arm of chromosome 7. It encodes a protein of 1,480 amino acids called the cystic fibrosis transmembrane conductance regulator (CFTR). A three-base deletion removing a phenylalanine residue at position 508 of CFTR (delta F508 mutation) is present on approximately 70% of CF chromosomes. The remaining cases are explained by more than 1,200 mutations, none of which accounts for more than 2% of the cases. The ability to detect CF mutations by direct DNA analysis allows for prenatal diagnosis, newborn screening, and heterozygote detection. Mutation analysis also can be used to confirm a diagnosis of CF. Genotype predicts pancreatic function, but, with rare exception, it does not predict the severity of pulmonary disease or overall clinical course. Evidence indicates that environmental factors and genes other than CFTR modify the severity of the disease.
PATHOPHYSIOLOGY
The CFTR protein is part of the ABC transporter family of membrane-bound glycoproteins involved in the transport of small molecules across cell membranes. CFTR functions as a cyclic adenosine monophosphate (cAMP)-activated chloride channel on the apical surface of epithelial cells and also regulates other membrane conductance pathways. The CFTR protein contains two nucleotide-binding folds with adenosine triphosphate-binding sites, a regulatory region with many phosphorylation sites, and two hydrophobic regions that probably interact with cell membranes. Functional expression of the CF defect reduces the ability of epithelial cells in the airways and pancreas to transport chloride in response to cAMP-mediated stimuli. Abnormal transport of sodium and chloride across the airway epithelium is thought to lead to water or volume-depleted periciliary fluid, increased viscosity of airway mucus, and the abnormal mucociliary clearance seen in patients with CF. Complementation of the CF defect has been accomplished in CF airway epithelial cells in vivo and in animal models using viral and cationic liposome vectors.
When first descibed, CF was thought to involve primarily the pancreas, but many of the clinical and pathologic findings can be explained by a generalized defect in mucus secretion. Discovery of the sweat gland defect made it apparent that abnormalities exist in all exocrine glands. Glands are affected in varying distribution and degrees of severity and fall into three types: those obstructed by viscid or solid eosinophilic material in the lumen (pancreas, intestinal glands, intrahepatic bile ducts, gallbladder, submaxillary glands), those that produce an excess of histologically normal secretions (tracheobronchial and Brunner glands), and those that are histologically normal but secrete excessive electrolytes (sweat, parotid, and small salivary glands). The high concentration of electrolytes in sweat is the result of decreased transductal reabsorption of chloride and sodium.
CLINICAL FEATURES
Box 236.1 is a summary of clinical features consistent with a diagnosis of CF.
Pulmonary System
The respiratory tract almost always is involved, and pulmonary complications usually dominate the clinical picture. However, manifestations may not appear until weeks, months, or even years after birth. Autopsy studies suggest that the lungs are normal at birth. The initial pulmonary lesion is obstruction of the small airways by abnormally thick mucus secretions. Secondary to obstruction, bronchiolitis and mucopurulent plugging of the airways occur. Bronchial changes are more common findings than are parenchymal changes. Bronchiectasis is present in almost all patients older than 18 months of age. It progresses with age and is especially striking in older patients. Emphysema is not a common finding. Figure 236.1 shows a proposed mechanism for the pathophysiology of lung disease in CF.
Infection
Secondary bacterial infection, caused initially by Staphylococcus aureus and then by Pseudomonas aeruginosa, initiates a cycle of airway obstruction, chronic infection and inflammation leading to tissue damage, and eventual ventilatory failure. More than 80% of patients with advanced disease consistently harbor strains of P. aeruginosa, most of which are heavy slime producers known as mucoid variants. These variants rarely are found in other diseases. The susceptibility of patients with CF to infection with mucoid Pseudomonas strains may be related to a defect in phagocytosis in the lung or increased adherence
of P. aeruginosa to respiratory tract receptors or mucins. The possibility of progression in the airways from intermittent colonization with nonmucoid strains to chronic colonization with nonmucoid strains and then chronic colonization with mucoid strains has been proposed. On initial acquisition, eradication of transient Pseudomonas may be possible, but once established in the airways, Pseudomonas is virtually impossible to eradicate. Systemic defense mechanisms appear to be intact, and infection tends to be localized to the respiratory tract. Septicemia and extrapulmonary infections are rare occurrences.
of P. aeruginosa to respiratory tract receptors or mucins. The possibility of progression in the airways from intermittent colonization with nonmucoid strains to chronic colonization with nonmucoid strains and then chronic colonization with mucoid strains has been proposed. On initial acquisition, eradication of transient Pseudomonas may be possible, but once established in the airways, Pseudomonas is virtually impossible to eradicate. Systemic defense mechanisms appear to be intact, and infection tends to be localized to the respiratory tract. Septicemia and extrapulmonary infections are rare occurrences.
BOX 236.1. Clinical Features Consistent with a Diagnosis of Cystic Fibrosis
Chronic Sinopulmonary Disease Manifestations
Persistent colonization or infection with typical cystic fibrosis pathogens, including Staphylococcus aureus, nontypeable Haemophilus influenzae, mucoid and nonmucoid Pseudomonas aeruginosa, and Burkholderia cepacia
Chronic cough and sputum production
Persistent chest radiograph abnormalities (e.g., bronchiectasis, atelectasis, infiltrates, hyperinflation)
Airway obstruction (wheezing and air trapping)
Nasal polyps; radiographic or computed tomographic abnormalities of the paranasal sinuses
Digital clubbing
Gastrointestinal and Nutritional Abnormalities
Intestinal: meconium ileus; distal intestinal obstruction syndrome; rectal prolapse
Pancreatic: pancreatic insufficiency; recurrent pancreatitis
Hepatic: chronic hepatic disease manifested by clinical or histologic evidence of focal biliary cirrhosis or multilobular cirrhosis
Nutritional: failure to thrive (protein-calorie malnutrition), hypoproteinemia and edema, complications secondary to fat-soluble vitamin deficiency
Salt Loss Syndromes (Acute Salt Depletion; Chronic Metabolic Alkalosis)
Male Urogenital Abnormalities Resulting in Obstructive Azoospermia
During the 1990s, the incidence of Burkholderia cepacia colonization among adolescent and adult patients was increased. Risk factors for colonization include increasing age and severity of underlying disease and recent hospitalization. Person-to-person transmission has been documented. Acquisition of this organism, especially by patients with moderate or advanced pulmonary disease, may be followed by an unexpectedly rapid decline. In some patients, the course is characterized by recurrent episodes of fever and bacteremia termed Cepacia syndrome. Colonization of the airways with methicillin-resistant S. aureus and with several gram-negative organisms, including Stenotrophomonas maltophilia and Achromobacter xylosoxidans, is seen in 5% to 10% of patients. If patients do not respond as expected to antimicrobial therapy, infection with unusual organisms such as Mycobacterium tuberculosis, nontuberculous mycobacteria (present in up to 10% of older patients), and Aspergillus should be considered.
Inflammatory Lung Damage
Inflammation contributes significantly to lung damage in patients with CF. Recruitment and activation of neutrophils in the airways lead to high levels of DNA, interleukin-8, and free neutrophil elastase in bronchoalveolar lavage fluid. Increased elastolytic activity secondary to the release of proteases is associated with breakdown of the lung matrix and with cleavage and inactivation of a variety of opsonins, thereby contributing to the persistence of Pseudomonas in the lung. An exaggerated and prolonged inflammatory response to bacterial and viral pathogens occurs. Significant lung inflammation has been documented in early infancy and in older patients before the onset of clinically apparent pulmonary disease. Immune complex formation is present in 20% to 100% of patients and may contribute to inflammatory lung damage. The presence of immune complexes correlates with disease severity and prognosis.
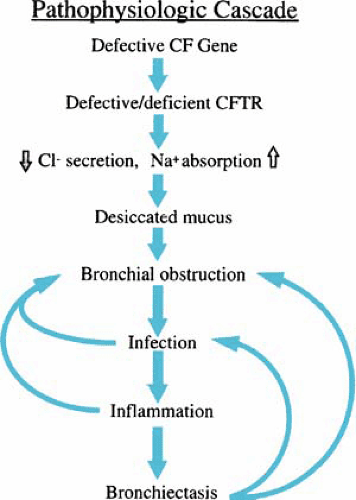 FIGURE 236.1. Proposed pathophysiologic cascade of cystic fibrosis lung disease. CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator. (Reprinted with permission from Davis, PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med 1986;154:1229.) |
Signs and Symptoms
Half of all patients present with pulmonary manifestations usually consisting of chronic cough and wheezing along with recurrent or chronic infections. Infants can present with atelectasis, often involving the right upper lobe, or a severe bronchiolitic syndrome. The most prominent and constant feature of pulmonary involvement is chronic cough. At first the cough may be dry, but with progression of disease, it becomes paroxysmal and productive. Older patients expectorate mucopurulent sputum, particularly in association with pulmonary exacerbations. Often, wheezing is a prominent feature, especially in association with pulmonary exacerbations, but whether it reflects inflammation and
bronchial obstruction or coincidental atopy remains unclear. As many as 5% of patients develop allergic bronchopulmonary aspergillosis. Physical findings include a barrel-chest deformity, kyphosis, use of accessory muscles of respiration, growth retardation, digital clubbing, pulmonary hypertrophic osteoarthropathy, and cyanosis.
bronchial obstruction or coincidental atopy remains unclear. As many as 5% of patients develop allergic bronchopulmonary aspergillosis. Physical findings include a barrel-chest deformity, kyphosis, use of accessory muscles of respiration, growth retardation, digital clubbing, pulmonary hypertrophic osteoarthropathy, and cyanosis.
Radiographic Changes
Chest radiographic and CT findings are not pathognomonic but can help to suggest a diagnosis of CF. Hyperinflation and bronchial wall thickening are the earliest findings. Subsequent changes include areas of infiltrate, atelectasis, and hilar adenopathy. With advanced disease, segmental or lobar atelectasis, cyst formation, bronchiectasis, and enlargement of the pulmonary arteries and right ventricle are seen. Characteristic branching, fingerlike opacifications representing mucoid impaction of dilated bronchi strongly suggest CF. In patients with mild disease and normal chest radiograph findings, bronchial wall thickening and bronchiectasis may be evident on high-resolution CT.
Pulmonary Function
Airway obstruction, air trapping, and ventilation-perfusion inequalities are the most important functional changes in CF. Usually, ventilation-perfusion scans demonstrate focal areas of inequality. Pulmonary function tests reveal the following: hypoxemia; reduction in forced vital capacity (FVC), in forced expiratory volume over 1 second (FEV1), and in the ratio of FEV1 to FVC; and an increase in residual volume and in the ratio of residual volume to total lung capacity. Flow-volume loops demonstrate a characteristic scooped-out appearance, indicative of small-airways disease (Fig. 236.2) and flow limitation at lower lung volumes. Airway reactivity, based on bronchoprovocative challenges, is present in 50% of patients and may be associated with accelerated progression of pulmonary disease. The response to bronchodilators is unpredictable and varies over the course of time and with changes in underlying pulmonary status.
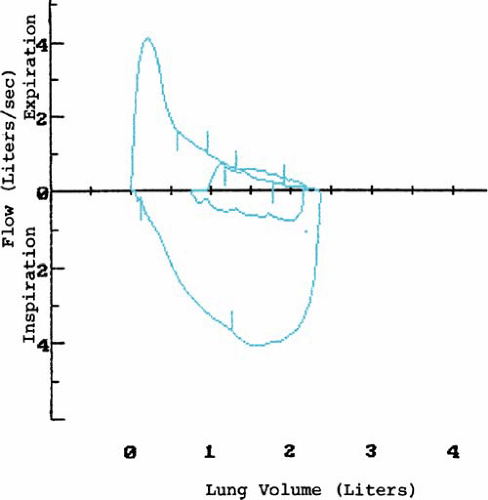 FIGURE 236.2. Flow-volume loop from a 23-year-old patient showing decreased flow at all lung volumes. The scooped-out appearance is indicative of small-airways disease. |
Pneumothorax
In patients with advanced lung disease, pneumothorax, hemoptysis, and cor pulmonale are frequent complications. Pneumothorax occurs secondary to rupture of subpleural blebs. The cumulative incidence is 2% to 10% and, in adults, may be as high as 16%. Typically, patients present with acute onset of chest pain and respiratory distress. Spontaneous pneumothorax generally is a poor prognostic sign. After having a pneumothorax on one side, the patient has an increased risk of having an pneumothorax on the contralateral side within 6 to 12 months, and the median survival is approximately 48 months.
Hemoptysis
Often, patients experience blood streaking of their sputum. Bleeding is caused by erosion of abnormal bronchial arteries into a bronchus, often in association with an exacerbation of the underlying pulmonary infection. Massive hemoptysis (greater than 240 mL/24 hours) occurs in 1% of cases each year and is associated with significant mortality and high recurrence rates. The site of bleeding may be suggested by chest radiograph or CT findings and may be localized by bronchoscopy.
Cor Pulmonale
Cor pulmonale, manifested by hypertrophy of the right ventricle, is seen in 70% of patients dying with CF and occurs in 50% of patients surviving past age 15 years. Chronic alveolar hypoxia and hypoxemia serve as stimuli to vasoconstriction and medial hypertrophy of the pulmonary arteries. Severe cor pulmonale is associated with PaO2 values of less than 50 mm Hg. Clinical recognition of cor pulmonale may be difficult. Peripheral edema is present in only two-thirds of cases and often is a late manifestation. Liver tenderness may be an early clue. Electrocardiography does not correlate consistently with the presence of right ventricular hypertrophy. Echocardiography is the most practical and reliable way of documenting cor pulmonale and of following its course.
Upper Respiratory Tract
The upper respiratory tract is affected secondary to hyperactive mucus-secreting glands and hyperplasia and edema of mucous membranes. Symptoms of sinonasal disease are common and include nasal obstruction, rhinorrhea, headache, and mouth breathing. Nasal polyps occur in 6% to 48% of patients. They occur at a much younger age in patients with CF compared with those with underlying atopy, can be differentiated histologically, and tend to recur. On examination, the patient may have congestion and erythema of the nasal mucosa, abundant secretions, and broadening of the nasal bridge. Radiography and CT may be helpful in diagnosing CF because almost all patients with CF have opacification of the paranasal sinuses. Other findings include lack of development or hypoplasia of the frontal and sphenoid sinuses, demineralization of the medial walls, destruction of the lateral walls of the maxillary sinuses, and formation of mucoceles. CT scans can be helpful in selecting patients for surgery. The microbial flora (including Pseudomonas) of the sinuses tends to mirror that of the lower respiratory tract.
These patients have an increased incidence of sensorineural hearing loss for high-frequency sounds consistent with aminoglycoside-induced ototoxicity.
Course
The pulmonary course is characterized by chronic suppurative endobronchial infection with recurrent pulmonary exacerbations, often after viral respiratory infections. Infection with respiratory syncytial virus may be an important cause of significant respiratory morbidity in young infants. By age 10 years, 90% of patients have intermittent sputum production; by age 15 years, 90% have daily sputum production. Progressive shortness of breath and exercise intolerance occur. Pulmonary involvement advances at a variable rate, usually faster in female than in male patients, and eventually leads to respiratory failure, cardiac failure, or both.
Gastrointestinal System
Pancreatic Exocrine Deficiency
The most common gastrointestinal manifestations result from loss of pancreatic enzyme activity and consequent intestinal malabsorption of fats, proteins and, to a lesser extent, carbohydrates. Complete loss of enzyme activity is seen in 85% to 90% of patients. Loss of function may be progressive. Clinical manifestations include the following: poor weight gain; abdominal distention; deficiency of subcutaneous fat and muscle tissue; rectal prolapse; and frequent passage of pale, bulky, malodorous, and often oily stools. Steatorrhea and azotorrhea may be pronounced. Secondary to pancreatic insufficiency, patients have low serum lipid levels and may be deficient in fat-soluble vitamins and linoleic acid. Although some patients have a voracious appetite, often caloric intake is deficient. In adolescent patients, absence of a pubertal growth spurt and delayed maturation may occur. In general, growth retardation correlates closely with the degree of pulmonary involvement. Patients with pancreatic sufficiency tend to have lower sweat chloride values, less severe pulmonary involvement, better nutritional status, and better survival. They may have episodes of pancreatitis, sometimes as their presenting manifestation of CF.
Evaluation of Pancreatic Function
Exocrine pancreatic function can be evaluated by direct and indirect methods. The most direct measure involves analysis of duodenal fluid before and after the intravenous injection of pancreozymin and secretin. In patients with CF, volume and bicarbonate secretion (ductular activity) are reduced grossly, regardless of the presence of steatorrhea. In patients with steatorrhea, enzyme secretion (acinar activity) is virtually absent. However, this procedure is technically difficult and generally is used only in research studies.
Indirect and less invasive measures of pancreatic exocrine function include serum immunoreactive trypsin (IRT) levels, 72-hour fecal fat excretion, stool levels of trypsin and chymotrypsin, and the fecal elastase-1 test. The elastase-1 test is particularly useful because a normal test result has 99% predictive value for ruling out pancreatic insufficiency.
Carbohydrate Intolerance
In addition to experiencing pancreatic exocrine dysfunction, as many as 40% of patients show carbohydrate intolerance that progresses to chronic insulin-requiring diabetes in 15% to 20% of patients older than 18 years of age. The average age of onset of diabetes is between 18 and 21 years, but it can occur at any age. Diabetes in patients with CF is characterized by insidious onset, mild clinical course, and virtual absence of ketoacidosis. Deterioration in nutritional and pulmonary status may occur several years before CF-related diabetes is diagnosed. Retinopathy, nephropathy, and neuropathy may occur, with an adverse effect on survival. The mild course in patients with CF may result from preservation of some insulin secretion, decreased glucagon release, and compensatory enhancement of peripheral tissue sensitivity to insulin. Beginning in adolescence, annual glucose tolerance testing should be performed.
Meconium Ileus
Meconium ileus (MI), which occurs secondary to obstruction of the distal ileum by inspissated, tenacious meconium, occurs in 18% of newborns with CF. The presence of echogenic bowel on a prenatal ultrasound scan may be an early clue to the diagnosis. With rare exception, MI always is associated with CF. MI most likely is related to in utero deficiency of proteolytic enzymes, along with secretion of abnormal mucoproteins by the goblet cells of the small intestine. Infants present with evidence of intestinal obstruction. Abdominal radiography shows distended bowel loops and a “bubbly” pattern of inspissated meconium in the terminal ileum. Contrast enema shows a microcolon from disuse secondary to intrauterine obstruction. Associated intestinal complications, including small bowel atresia, volvulus, and perforation or peritonitis, are present in 40% to 50% of cases. MI tends to recur in patients’ families. A delay in the passage of meconium and distal colonic obstruction secondary to the meconium plug syndrome also may be presenting manifestations of CF.
Late Intestinal Complications
The intestinal contents tend to be abnormally thick and puttylike as a result of abnormal intestinal gland secretions, abnormal chloride and water movement across the colonic epithelium, deficiency of pancreatic enzymes, and prolonged intestinal transit time. This condition may lead to a variety of late intestinal complications. Recurrent episodes of partial or complete obstruction of the small or large bowel, often preceded or accompanied by crampy abdominal pain, distention, anorexia, and a palpable mass in the right lower quadrant may occur. This symptom complex is called distal intestinal obstruction syndrome (DIOS) and occurs in as many as 15% of patients. Episodes may be precipitated by dehydration, change in diet, or inadequate enzyme supplementation. The incidence of severe obstruction in the immediate postoperative period following lung transplantation is high. Precipitated by the abnormal intestinal contents, episodes of small bowel volvulus or intussusception also may occur. The latter complication occurs in 1% of older patients and may be the presenting manifestation of CF. Episodes tend to recur and may be associated with chronic symptoms.
The diagnosis of CF can be suggested by the histologic features of the appendix. Goblet cells are increased in number and are distended with mucus, and eosinophilic casts may fill the crypts and extend into the lumen. The incidence of acute appendicitis probably is lower in patients with CF as compared with the general population. However, often the diagnosis is delayed because of an atypical or subacute presentation and confusion with DIOS. Development of a chronic abdominal abscess related to unrecognized appendicitis sometimes is seen. Mucoid impaction of the appendix may present as a right lower quadrant mass in the absence of other symptoms.
Recurrent episodes of rectal prolapse occur in as many as one-fourth of patients, most often before the diagnosis is established. Rectal prolapse probably is related to frequent bulky stools, malnutrition, and raised intraabdominal pressure secondary to paroxysmal cough. The diagnosis of CF should be considered in every patient with rectal prolapse.
Upper Gastrointestinal Complications
Patients with CF have an increased prevalence of gastroesophageal reflux, probably related to chest hyperinflation along with increased abdominal pressure resulting from coughing. This disorder may be manifested by regurgitation and failure to thrive in infants and by dysphagia, epigastric pain, esophageal ulcerations, and blood loss in older patients. Diagnosis is confirmed by prolonged pH monitoring, endoscopy, and biopsy. Despite increased gastric acid secretion and a low pH in the duodenum (secondary to decreased pancreatic bicarbonate output), duodenal ulcers infrequently have been diagnosed antemortem. A high incidence of peptic ulcer disease, however, has been reported in black adolescents. Radiographically, duodenal abnormalities consisting of thickened
mucosal folds, nodular filling defects, and mucosal smudging are present in 80% of patients. Because of these abnormalities, diagnosing peptic ulcer disease radiographically may be difficult. In patients with signs and symptoms suggestive of peptic ulcer disease, endoscopy is the preferred diagnostic procedure.
mucosal folds, nodular filling defects, and mucosal smudging are present in 80% of patients. Because of these abnormalities, diagnosing peptic ulcer disease radiographically may be difficult. In patients with signs and symptoms suggestive of peptic ulcer disease, endoscopy is the preferred diagnostic procedure.
Miscellaneous Gastrointestinal Complications
Crohns disease, giardiasis, Clostridium difficile-associated colitis, and celiac disease have been reported in patients with CF, and evidence suggests that patients may be at increased risk for developing these conditions. A significantly increased risk of development of digestive tract (colon, small intestine, biliary tract) cancers exists in adult patients. The risk is even more pronounced among patients who have had an organ transplant. This possibility must be considered in adult patients with intestinal obstruction, chronic abdominal pain, and gastrointestinal bleeding.
Nutrition and Metabolism
Vitamin and Mineral Deficiencies
Secondary to pancreatic achylia, malabsorption of fat-soluble vitamins occurs. Low-serum vitamin A levels result from steatorrhea and a reduction of retinol-carrier protein and retinol-binding protein. Xerophthalmia and night blindness occur rarely, usually in association with hepatic involvement. A bulging fontanelle secondary to vitamin A deficiency may be the presenting manifestation in infants. Overt rickets is a rare finding, but a significant reduction in vitamin D biologic activity with associated secondary hyperparathyroidism, reduced bone mineral content, and delayed bone maturation is seen commonly. Significant demineralization is present in half of all patients. Severe bleeding in association with hypoprothrombinemia and deficiency of clotting factors II, VII, IX, and X secondary to vitamin K deficiency may occur in infants. Hypoprothrombinemia is especially likely to occur in association with hepatic involvement or prolonged antibiotic administration. All patients with pancreatic achylia show a marked reduction in plasma alpha-tocopherol levels. Histologic evidence of vitamin E deficiency consists of focal necrosis of striated muscle and ceroid pigment deposition in intestinal smooth muscle. Red blood cells show a moderate decrease in survival that, on occasion, may result in hemolytic anemia. A progressive spinocerebellar syndrome consisting of ataxia, areflexia, and proprioceptive loss is seen in patients with prolonged vitamin E deficiency. Clinical problems secondary to deficiency of water-soluble vitamins are rare occurrences, but angular stomatitis may develop secondary to riboflavin deficiency.
Symptomatic hypomagnesemia has been reported in patients receiving prolonged aminoglycoside therapy and in those being treated for DIOS. Low plasma levels of zinc have been reported in 30% of young infants identified by newborn screening, and infants may present with signs and symptoms (acrodermatitis enteropathica) of severe zinc deficiency. Selenium levels may be low but do not correlate with clinical manifestations of deficiency. Iron deficiency anemia is seen in one-third of patients. Probably, it is related to inadequate iron intake, impairment of iron absorption, and chronic infection.
Edema and Hypoproteinemia
The syndrome of edema and hypoproteinemia, secondary to pancreatic enzyme deficiency, may be the presenting manifestation in as many as 8% of patients. Most often, it is seen in infants who are between ages 1 to 6 months and are breast-fed or receiving soy-based formula. Associated findings include hepatomegaly, elevation of liver enzymes, skin rash (acrodermatitis enteropathica), and anemia. False-negative sweat test results can be seen in the presence of edema.
Salt Loss
The sweat gland abnormality has important clinical implications. Patients may have a “salty taste” or salt crystal formation on the skin, findings that are highly suggestive of CF. Patients also may develop dehydration with massive salt depletion, especially in association with gastrointestinal losses or thermal stress. Profound hypoelectrolytemia, not accounted for by gastrointestinal losses, particularly suggests CF. In arid climates, chronic salt loss can lead to metabolic alkalosis and depletion of electrolytes without appreciable dehydration and is a common presenting manifestation of CF.
Hepatobiliary System
Liver
The liver is involved extensively in CF, and in some patients, liver complications may be the predominant and, at times, presenting features. Liver complications, which are seen almost exclusively in patients with pancreatic insufficiency, may have a familial pattern of occurrence.
Focal biliary cirrhosis, characterized by the inspissation of amorphous, granular material in the intrahepatic bile ducts, bile duct proliferation, inflammatory infiltrate, a variable degree of fibrosis, and focal distribution, is pathognomonic of CF. It is associated with and probably caused by an excessive accumulation of biliary mucus. Release of inflammatory mediators in the obstructed bile ducts leads to progressive liver damage. This lesion is present in 25% of patients and may appear as early as age 3 days. Usually, it produces no clinical manifestations, but, secondary to intrahepatic bile stasis, prolonged neonatal jaundice may be present. Half of such cases occur in association with MI and may predispose to later liver complications. In 4% to 6% of patients, focal biliary cirrhosis progresses to multilobular biliary cirrhosis that consists of groups of normal-appearing lobules surrounded by dense fibrous septa containing proliferating bile ducts with eosinophilic concretions. This lesion is specific for CF. Features of primary sclerosing cholangitis may be present in one-third of adult patients. Portal hypertension manifested by esophageal varices and hypersplenism develops in 1% to 2% of all patients, with a peak incidence of 2.7% in those between ages 16 and 20 years. Thrombocytopenia may be an early clinical clue. Liver failure is a rare finding. CF-related liver disease usually is slowly progressive and may remain asymptomatic over many years. The diagnosis often is problematic in that many patients have elevations of liver enzymes and ultrasound abnormalities in the absence of significant liver disease, whereas others have only mild enzyme elevations in the presence of severe cirrhosis. Liver biopsy results may be helpful but can be misleading because of the focal nature of the process.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


