Fig. 10.1
Annual changes in motor function. Right column, ambulant patients; left column, nonambulant patients. a 6MWT; b, c summed MMT; d, e grip power; and f, g %FVC. All patients, with the exception of one (*), showed deterioration in 6MWT. (a) Only one patient with improved 6MWT succeeded in weight control and had more opportunities to walk relative to baseline. Both ambulant (b) and nonambulant (c) patients showed deterioration in summed MMT. The decrease in grip power was greater in ambulant patients (d, e), whereas the decrease in %FVC was greater in nonambulant patients
10.1.2.4 Limitations and Conclusions
Summed MMT, grip power, and %FVC significantly changed over the course of a year. Although results of the 6MWT were not significant, which likely reflects the small number of ambulant patients, a larger cohort may clearly detect the deterioration. The 6MWT and summed MMT are important end-point item candidates for clinical trials because they can be used to determine annual changes in disease progression. Our study showed reductions in respiratory function, especially among nonambulant patients, suggesting that %FVC is a useful outcome measure for nonambulant patients. Our data suggest that GNE myopathy does not involve cardiomyopathy, although cardiac involvement was previously implicated in a mouse model [11].
Limitations include the small number of patients and short study period. In rare diseases such as GNE myopathy, large-scale studies tend to be difficult.
In conclusion, 6MWT, summed MMT, GMFM, grip power tests, and %FVC may be good clinical evaluation tools for clinical trials and to correlate with disease progression, although %FVC and grip power should be used according to ambulation status.
10.1.3 Patient Registry Development
10.1.3.1 Aims
Our previous studies made clear the limitations of natural history studies, which involved small sample populations. In order to gain further insight into GNE myopathy for the purpose of improving therapy and care, more patient data are required. To this end, we aimed to develop a nationwide patient registry for GNE myopathy in order to facilitate the planning of clinical trials and recruitment of candidates and to disseminate standard care and current information to patients, physicians, and researchers.
10.1.3.2 Study Design
Medical records of genetically confirmed patients with GNE myopathy at the NCNP were retrospectively reviewed in order to obtain data reflecting the severity and progression of the disease. Items selected for the registration sheet included age, sex, age at onset, past history and complications, family history, body weight and height, pathological findings from muscle biopsy, grip power, walking ability, respiratory function, cardiac function, willingness to join upcoming clinical trials, and participation in patient associations. A copy of the original genetic analysis report was required of each patient.
10.1.3.3 Institution, Organization, Registration Method, Data Collection, and Ethical Approval
In 2009, we developed a national registry for neuromuscular diseases (Registry of Muscular Dystrophy, Remudy; http://www.Remudy.jp/, see also Chap. 11) in Japan in collaboration with the TREAT-NMD Alliance in order to aid in the recruitment of eligible patients for clinical trials, provide information regarding the natural history and epidemiology of diseases, and serve as a source of information on current clinical care [13]. Remudy is supported by Intramural Research Grants (23-4/26-7) for Neurological and Psychiatric Disorders from the NCNP. Registry information was provided to interested individuals and their informed consent was obtained. Individuals whose data were included were informed that inclusion in the database confers no obligation to the patient and that they will be removed from the registry immediately upon request. They were also told that refusal to participate would not affect subsequent medical care. Study objectives, design, risks, and benefits of participation were explained to all patients, and their written informed consent was obtained prior to enrollment.
10.1.3.4 Results
As of the end of October 2013, a total of 121 Japanese patients with GNE myopathy (55 men and 66 women) had registered. Mean ages at data collection and disease onset were 44.9 ± 13.2 years (median, 43 years; range, 21–85 years) and 27.9 ± 9.6 years (median, 26 years; range, 12–61 years), respectively. The registry included patients from throughout Japan (38/47 prefectures) who were recruited through a collaboration with 92 attending physicians from 73 institutes (Fig. 10.2). Three patients had a past history of idiopathic thrombocytopenia (ITP).
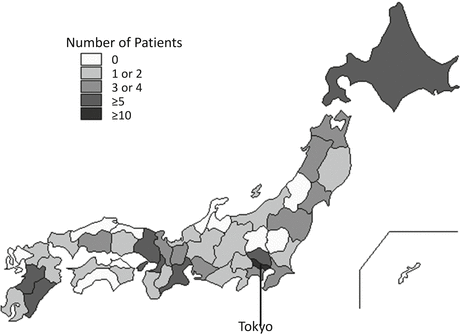
Fig. 10.2
Patient distribution. Patients were distributed throughout Japan (38/47 prefectures), and 92 physicians at 73 institutes agreed to contribute to the registry
Thirty-nine of 121 patients (32.3 %) harbored a homozygous mutation in GNE, and 78 of 121 (64.5 %) had a compound heterozygous mutation. Only one heterozygous mutation was found in four (3.3 %) patients. Among those with a homozygous mutation, 82 % (32/39), 8 % (3/39), and 5 % (2/39) harbored p. V572L, p. C13S, and p. M172T mutations, respectively. A homozygous mutation of p. D176V was identified in only one patient. Of those carrying two heterozygous mutations, 31 % (24/78) had p. D176V/p. V572L mutations, while the remaining patients had other combinations of mutations.
Mean age at disease onset was 27.7 ± 9.6 years (median, 27.5 years; interquartile range, 15–61), 20 % (24/121) were ambulant without assistance, 37 % (45/121) required assistance (e.g., canes and/or braces), and 43 % (52/121) had lost ambulation. Mean age at loss of ambulation was 35.4 ± 11.3 years. Kaplan–Meier analysis revealed median durations from disease onset to walking with assistance, wheelchair use, and loss of ambulation of 8.9 years (95 % CI, 6.3–9.7), 14.0 years (95 % CI, 11.8–16.2), and 21.0 years (95 % CI, 15.4–26.6), respectively.
Related posts:
 Toward Regenerative Medicine for Muscular Dystrophies
Therapeutic Approach of iPS Cell Technology for Treating Muscular Dystrophy
Toward Regenerative Medicine for Muscular Dystrophies
Therapeutic Approach of iPS Cell Technology for Treating Muscular Dystrophy
 Molecular Pathogenesis and Therapeutic Strategy in GNE Myopathy
Molecular Pathogenesis and Therapeutic Strategy in GNE Myopathy
 Stem Cell-Based Therapy for Duchenne Muscular Dystrophy
Stem Cell-Based Therapy for Duchenne Muscular Dystrophy
 Translational Research on DMD in Japan
Translational Research on DMD in Japan
 Patient Registries for International Harmonized Clinical Development
Patient Registries for International Harmonized Clinical Development
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


