For diagnostic evaluation of a neuromuscular disease, the clinician must be able to obtain a relevant patient and family history and perform focused general, musculoskeletal, neurologic, and functional physical examinations to direct further diagnostic evaluations. Laboratory studies for hereditary neuromuscular diseases include the relevant molecular genetic studies. The electromyogram and nerve-conduction studies remain an extension of the physical examination, and help to guide further diagnostic studies such as molecular genetics and muscle and nerve biopsies. All diagnostic information needs are to be interpreted within the context of relevant historical information, family history, physical examination, laboratory data, electrophysiology, pathology, and molecular genetics.
- •
Progressive acquired or hereditary neuromuscular diseases (NMDs) are disorders caused by an abnormality of any component of the lower motor neuron: anterior horn cell, peripheral nerve, neuromuscular junction (presynaptic or postsynaptic region), or muscle.
- •
Many NMDs are multisystem disorders affecting multiple organ systems.
- •
In the context of diagnostic evaluation of NMD, the clinician still must be able to obtain a relevant patient and family history and perform focused general, musculoskeletal, neurologic, and functional physical examinations to direct further diagnostic evaluations.
- •
Laboratory studies often include relevant molecular genetic studies in certain instances; however, specific genetic entities need to be strong diagnostic considerations, as these studies may be expensive and with limited sensitivity.
- •
Early diagnosis is facilitated by knowledge of the common initial clinical presentations of specific NMDs, and in many cases the early diagnosis has potential implications for treatment and prevention of secondary conditions.
Introduction
Progressive acquired or hereditary neuromuscular diseases (NMDs) are disorders caused by an abnormality of any component of the lower motor neuron: anterior horn cell, peripheral nerve, neuromuscular junction (NMJ; presynaptic or postsynaptic region), or muscle. The notion that a pathologic abnormality in an NMD may be purely isolated to one anatomic region of the lower motor neuron with primary or secondary changes isolated to muscle is only true for selected conditions. Many NMDs are multisystem disorders affecting multiple organ systems. For example, RNA toxicity generated from expansion of trinucleotide repeat sequences in myotonic muscular dystrophy (MMD) gives rise to skeletal muscle, smooth muscle, myocardial, endocrine, brain, and ocular abnormalities; Duchenne muscular dystrophy (DMD) gives rise to abnormalities of skeletal and cardiac muscle, the cardiac conduction system, smooth muscle, and the brain; Fukuyama congenital muscular dystrophy affects skeletal muscle and brain; mitochondrial encephalomyelopathies may affect the mitochondria of multiple tissues.
The most common NMDs are acquired peripheral neuropathies. Other acquired NMDs include amyotrophic lateral sclerosis (ALS), poliomyelitis, Guillain-Barré syndrome, myasthenia gravis, and polymyositis. Hereditary NMDs are also quite common and include such disorders as spinal muscular atrophy (SMA), Charcot-Marie-Tooth disease (CMT), congenital myasthenia, and DMD. Clinical NMD syndromes described over the decades in the literature have recently been redefined based molecular genetic advances and documentation of genetic heterogeneity within specific syndromes. For example, at least 70 genetically distinct subtypes of CMT have been described, some with undetermined gene loci; more than 14 genetically distinct subtypes of autosomal recessive limb-girdle muscular dystrophy (LGMD) have been identified; and 3 genetically distinct subtypes of Emery-Dreifuss muscular dystrophy exist. In fact, the gene loci for more than 500 distinct neuromuscular and mitochondrial disorders have been identified at the time of writing. The basis for the use of molecular genetic studies for diagnosis is well described by Arnold and Flanigan in their article elsewhere in this issue.
In the context of diagnostic evaluation of NMD, the clinician still must be able to obtain a relevant patient and family history and perform focused general, musculoskeletal, neurologic, and functional physical examinations to direct further diagnostic evaluations. Laboratory studies often include relevant molecular genetic studies in certain instances; however, specific genetic entities need to be strong diagnostic considerations, as these studies may be expensive and with limited sensitivity.
Electrodiagnostic studies including electromyography (EMG) and nerve conduction studies remain an extension of the physical examination and help to guide further diagnostic evaluation such as molecular genetic studies (as in the case of CMT), muscle and nerve biopsies, or even motor point biopsies applied to the evaluation of congenital myasthenic syndromes. All diagnostic information needs to be interpreted not in isolation, but within the context of relevant historical information, family history, physical examination findings, laboratory data, electrophysiologic findings, and pathologic information if obtained.
A skilled synthesis of all available information may provide the patient and family with: (1) a precise diagnosis or as accurate a diagnosis as is medically possible; (2) prognostic information (if available for a specific entity); (3) information as to eligibility for molecular based therapeutic agents such as antisense oligonucleotides or morpholinos for exon skipping, or stop-codon read-through agents; and (4) anticipatory guidance for the near future. Knowledge of the natural history of specific NMD conditions helps in the ongoing rehabilitative management of progressive impairments, activity limitations, and disabilities.
This article briefly reviews the clinical approach to the diagnostic evaluation of progressive NMDs, including relevant history, family history, clinical examination findings, laboratory studies, and situations whereby pathologic studies play a role diagnostically.
Neuromuscular disease history
Important elements of NMD history taking are shown in Box 1 .
- •
Distribution of Weakness
- ○
Anatomic distribution/pattern of weakness and focal wasting or hypertrophy of muscle groups (arms versus legs, proximal versus distal, symmetric versus asymmetric)
- ○
Myopathies have weakness that is usually proximal greater than distal with rare exceptions
- ○
- •
Course of weakness
- ○
Acute onset (days to weeks)
- ○
Chronic (months to years)
- ○
Episodic
- ○
Is the weakness getting worse, staying the same, or getting better?
- ○
Ascertain the rate of progression (days, weeks, months, or years)
- ○
- •
Fatigue or lack of endurance
- •
Muscle cramps or stiffness
- •
Lack of sensory loss
- •
Gait characteristics
- ○
Toe walking, excessive lordosis, Trendelenburg or gluteus maximus lurch, and so forth
- ○
- •
Functional difficulties
- ○
Ambulatory distances
- ○
Frequency of falls
- ○
Transitions from the floor to standing
- ○
Problems climbing stairs
- ○
Problems dressing
- ○
Problems reaching overhead
- ○
Difficulty lifting
- ○
Running ability, problems in physical education, and recreational or athletic performance
- ○
- •
Onset age (neonatal, childhood, teen, adult [20–60 years], or geriatric)
- •
Identify factors that worsen or help primary symptoms
- •
History of recent illnesses (eg, recent viral illnesses, respiratory difficulties, pneumonia, pulmonary infections)
- •
Pain
- •
Feeding difficulties, dysphagia, nutritional status, and body composition
- •
Cardiac symptoms (dizziness, syncope, chest pain, orthopnea, cardiac complaints with exertion)
- •
Pulmonary symptoms (breathing difficulties, sleep disturbance, morning headaches)
- •
Anesthetic history (eg, malignant hyperthermia)
- •
History regarding the child’s acquisition of developmental milestones
- ○
Ascertain when the child was able to control his or her head, sit independently, crawl, stand with and without support, walk with and without support, gain fine motor prehension, and acquire bimanual skills (bringing objects to midline, transfer of objects)
- ○
History regarding language acquisition, mental development and school performance
- ○
- •
History regarding pregnancy and neonatal period
- •
Quality of fetal movement, pregnancy complications, perinatal complications, evidence of fetal distress, respiratory difficulties in the recovery room, need for resuscitation or ventilation problems in early infancy, infantile hypotonia, weak cry, poor feeding
The common presenting chief complaints from parents of children with suspected neuromuscular disorders may include infantile floppiness or hypotonia, delay in motor milestones, feeding and respiratory difficulties, abnormal gait characteristics, frequent falls, difficulty ascending stairs or arising from the floor, muscle cramps, or stiffness. Adults often present with chief complaints of strength loss, fatigue or decreasing endurance, falls, difficulty ascending stairs, exercise intolerance, episodic weakness, muscle cramps, focal wasting of muscle groups, breathing difficulties, or bulbar symptoms relating to speech and swallowing.
Respiratory failure due to NMD has been reported in myasthenia gravis, myosin-loss myopathy, acid maltase deficiency, amyloidosis, desmin polymyositis (Jo-1), congenital myopathy (eg, rod; centronuclear), hydroxychloroquine toxicity, neural injury (specifically phrenic lesions), ALS, DMD, and SMA. NMDs with associated cardiac disorders include DMD, BMD, LGMD 1B, LGMD 1C, sarcoglycanopathies, MMD; McLeod, Emery-Dreifuss, Barth syndromes; desmin deficiency; Polymyositis; nemaline rod myopathy; Acid maltase deficiency; debrancher enzyme deficiency; carnitine deficiency; some mitochondrial myopathies; amyloid deficiency; drug-related disorders (metronidazole, emetine, and chloroquine, clofibrate, colchicine); cardiomyopathies; and some periodic paralyses.
Information should be obtained regarding the recent course of the chief complaint, specifically whether the process is getting worse, staying the same, or getting better. If strength is deteriorating, it is important to ascertain the rate of progression (ie, is weakness increasing over days, weeks, months, or years?). It is critical to determine whether the distribution of weakness is predominantly proximal, distal, or generalized. It is also useful to identify factors that may worsen or help the primary symptoms. A history of twitching of muscles may reflect fasciculations. Tremor or balance problems may be due to distal weakness or superimposed cerebellar involvement.
Bulbar involvement may be identified if the individual has difficulty chewing, swallowing, or articulating speech. Visual complaints (blurriness or diplopia) may indicate presence of cataracts or possibly involvement of extraocular musculature. Distal stocking glove or focal sensory complaints may be consistent with a peripheral neuropathy or focal nerve entrapment.
A comprehensive medical history and surgical history should be obtained. A history of recent illnesses should be carefully elucidated, including respiratory difficulties, aspiration pneumonias, or recurrent pulmonary infections. In addition, cardiac symptoms, such as dizziness, syncope, chest pain, orthopnea, or exertional cardiac complaints may indicate superimposed involvement of the myocardium. A pulmonary review of symptoms should be obtained. A history of weight loss may be due to recurrent illnesses, nutritional compromise, swallowing difficulty, or progressive lean tissue atrophy.
For the pediatric patient, a detailed history regarding pregnancy (eg, quality of fetal movement or pregnancy complications) and perinatal problems (evidence of fetal distress, respiratory difficulties in the delivery room, need for resuscitation or ventilation problems in early infancy, ongoing respiratory difficulties, swallowing/feeding difficulties, and persistent hypotonia) should be obtained. Perinatal respiratory distress in the delivery room may be seen in acute infantile type I SMA, myotubular myopathy, congenital hypomyelinating neuropathy, congenital infantile myasthenia, transitory neonatal myasthenia, and severe neurogenic arthrogryposis.
In children, history regarding the acquisition of developmental milestones should be ascertained relating to head control, independent sitting, crawling, standing with and without support, walking with and without support, fine prehension, bimanual skill acquisition (bringing objects to midline, transfer of objects), and language acquisition. Information regarding gait characteristics (toe walking, excessive lordosis, and so forth), running ability, transitions from floor to standing, stair climbing, falls, recreational/athletic performance, pain or muscle cramps, and easy fatigue or lack of endurance may be important clues to the presence of a neuromuscular disorder. History regarding mental development, type of school, and school performance may be important indicators of superimposed central nervous system (CNS) involvement.
For the adult, detailed history regarding the age of onset of symptoms, age when bracing was provided to maintain ambulation, age to use of wheelchair (if applicable), pattern of progression, distribution of weakness, presence of muscle cramps, fatigue, or episodic weakness, presence of atrophy or fasciculations, performance in physical education, military or vocational performance and pursuits, current and past ambulatory distances, ability to transition from floor to standing, problems climbing stairs, and problems reaching overhead or dressing may all be important functional information.
Potential causes of muscle cramps are shown in Box 2 . Muscle cramps in the setting of an elevated creatine kinase (CK) value and no skeletal muscle weakness has been reported, and a pedigree with mild Becker muscular dystrophy. The etiology of myalgias can be quite varied, and a definitive cause is found in only one-fourth of those patients presenting with muscle pain as a chief complaint. Patterns of weakness in myopathies, NMJ disorders, anterior horn cell disorders, and diagnostic considerations are outlined in Box 3 , and selective anatomic distribution of peripheral neuropathies and neuronopathies are listed in Box 4 .
- 1.
Cramps at rest (usually not a neuromuscular disorder)
- a.
Benign nocturnal leg cramps
- b.
Diurnal cramps related to exercise
- a.
- 2.
Cramps occurring with exertion, relieved by rest (may be associated with myoglobinuria)
- a.
Muscular dystrophy, Duchenne, Becker, limb-girdle muscular dystrophy (LGMD)
- b.
Myopathy: rippling muscle syndromes (RMD)
- i.
RMD1: chromosome 1q41; dominant
- ii.
RMD2: caveolin-3; chromosome 3p25.3; dominant
- i.
- c.
Metabolic disorders
- i.
Glycogenoses
- 1.
Myophosphorylase deficiency (type V; McArdle disease)
- 2.
Phosphofructokinase deficiency (type VII)
- 3.
Phosphorylase b kinase deficiency (type VIII)
- 4.
Phosphoglycerate kinase deficiency (type IX)
- 5.
Phosphoglycerate mutase deficiency (type X)
- 6.
Lactate dehydrogenase deficiency (type XI)
- 7.
Myoadenylate deaminase deficiency
- 1.
- ii.
Lipid metabolism disorders
- 1.
Carnitine palmityl transferase deficiency
- 1.
- iii.
Uremia
- iv.
Electrolyte abnormality: hyponatremia, hypocalcemia, hypomagnesemia, hypoglycemia
- v.
Hypothyroidism
- vi.
Hypoadrenalism
- vii.
Paroxysmal myoglobinuria
- viii.
Idiopathic rhabdomyolysis
- i.
- d.
Toxic myoglobinuria
- i.
Alcohol
- ii.
Barbiturates
- iii.
Heroin
- iv.
Carbon monoxide
- v.
Amphotericin B
- vi.
Toxic venoms
- i.
- e.
Inflammatory myositis
- i.
Acute dermatomyositis, polymyositis
- ii.
Viral myositis (Coxsackie and so forth)
- iii.
Bacterial myositis (staphylococci, clostridia)
- i.
- f.
Acute extracellular volume depletion
- i.
Perspiration
- ii.
Diarrhea, vomiting
- iii.
Diuretic therapy
- iv.
Hemodialysis
- i.
- g.
Other lower motor neuron disorders
- i.
Amyotrophic lateral sclerosis (ALS), old polio, other motor neuron disorders
- ii.
Radiculopathy and neuropathy
- i.
- a.
Extraocular Muscles (EOM) Weak
- •
Myasthenia gravis (MG)
- •
Thyroid
- •
Botulism
- •
Mitochondrial: Kearns-Sayre; progressive external ophthalmoplegia (PEO); MNGIE
- •
Myopathy: centronuclear; multicore
- •
Oculopharyngeal muscular dystrophy
- •
Inclusion body myositis (IBM) + contracture
- •
Oculopharyngodistal myopathy
- •
Congenital ophthalmoplegias
Periocular Without EOM Weakness
- •
Dystrophies: myotonic; facioscapulohumeral muscular dystrophy (FSHD); oculopharyngeal
- •
NMJ: MG
- •
Congenital myasthenic syndromes
- •
Congenital myopathies
- •
Inflammatory myopathy: polymyositis
- •
Rule out: VII nerve lesion
Bulbar Dysfunction
- •
NMJ: MG; congenital myasthenic syndromes
- •
Thyroid
- •
Cranial nerve Δ
- •
Oculopharyngeal muscular dystrophy
- •
Distal myopathy (MPD2)
- •
Polymyositis: IBM; scleroderma
- •
Motor neuron Δ: ALS; bulbospinal muscular atrophy (BSMA)
- •
Pseudobulbar; Fazio-Londe
- •
Brown-Vialetto-van Laere
Posterior Neck Weak
- •
Common: MG; polymyositis; ALS
- •
Focal myopathy: neck; paraspinous
- •
Rare: FSHD; LMN (lower motor neuron) syndrome; IBM rod; proximal myotonic myopathy (PROMM); acid maltase deficiency; hypo K + ; carnitine deficiency; endocrine deficiency; desmin deficiency
Proximal Arms Weak
- •
Dystrophy: scapuloperoneal; FSHD
- •
Inflammatory: brachiocervical inflammatory myositis (BCIM)
- •
Absent muscles; Shoulder joint Δ
- •
NMJ: MG
- •
Neuropathic: ALS; pure LMN
- •
Brachial plexopathy
Distal and Proximal Weakness
- •
Dystrophy: myotonic; FSH, scapuloperoneal
- •
Myopathy: congenital; distal
- •
Glygogenoses: debrancher
- •
Phosphorylase b kinase
- •
Neuropathy + Myopathy: paraneoplastic; sarcoid; mitochondria; human immunodeficiency virus (HIV)
- •
Drugs (amiodarone; doxorubicin; colchicine; chloroquine)
Acute Weakness
- •
NMJ: MG
- •
Myoglobinuria
- •
Myosin-loss myopathy
- •
Carnitine deficiency
- •
Periodic paralysis: X-episodic Xp22
- •
Hypo K + : CACNA1S; SCN4A; KCNE3
- •
Hyper K + : SCN4A; KCNE3
- •
Andersen: KCNJ2
- •
Electrolyte disorders: Hyperkalemia, hypokalemia, hypermagnesemia or hypophosphatemia
- •
Barium
- •
Rule out: Neuropathy (acute inflammatory demyelinating polyradiculoneuropathy [AIDP]), chronic inflammatory demyelinating polyradiculoneuropathy [CIDP]); spinal cord
Wasting > Weakness
- •
Pathology: type II atrophy
- •
Cachexia: weight loss >15%, aging/sarcopenia
- •
Disuse
- •
Steroid myopathy
- •
Paraneoplastic
Weakness > Wasting
- •
Polymyositis
- •
Myoglobinuria
- •
Periodic paralysis
- •
NMJ: MG; Congenital myasthenia
- •
Neuropathy + conduction block
Quadriceps Weak
- •
LGMD: 1B; 2B; 2H; ring fiber
- •
Becker
- •
Myositis: IBM; mitochondria; focal
- •
Nerve: femoral; lumbosacral plexopathy
- •
Diabetic amyotrophy; L3-L4 root
Extraocular Muscle
- •
Botulism
- •
Diabetes
- •
Miller-Fisher
- •
Diphtheria
- •
Rule out: MG; myopathy
Proximal Motor
- •
Immune demyelinating: Guillain-Barré syndrome; CIDP; SMA; porphyria
- •
Plexopathy: brachial; lumbar
- •
Rule out: joint pain; myopathy
Proximal Sensory
- •
Hereditary: porphyria; Tangier
- •
Neuronopathy: Hu; Sjögren; thoracic neuropathy
- •
Rule out: myelopathy
Skin Temperature–related
- •
Leprosy
- •
Upper extremity
- •
Immune: multifocal motor neuropathy (MMN); vasculitis; CIDP variant
- •
Amyloid: Carpal tunnel
- •
Entrapment: hereditary neuropathy with liability to pressure palsies (HNPP); other
- •
Toxic: lead; vincristine; ALS; LMN
- •
Rule out: spinal; CNS
Asymmetric
- •
Mononeuritis multiplex
- •
Neuronopathy: ALS; sensory
- •
Entrapments
- •
Plexopathies
- •
Toxic
Mononeuritis Multiplex
- •
Vasculopathy
- •
Amyloid
- •
Leprosy
- •
Diabetes
- •
Cytomegalovirus
- •
Waldenström
- •
Perineuritis
- •
Demyelinating: HNPP; multifocal CIDP; MMN
- •
Compression: multiple
- •
Lymphoma: intraneural
- •
Wartenberg
CNS
- •
Spinal: organophosphate; hexacarbon; Adrenomyeloneuropathy (AMN); metachromatic leukodystrophy (MLD); lymphoma; Cuban; Vernant
- •
Optic: disulfiram; CS2; Hg; drugs; Neuropathy, ataxia, and retinitis pigmentosa (NARP); Charcot Marie Tooth 6 (CMT6); Post col & Retinitis pigmentosa (RP); Cuban; Vernant
- •
Hearing loss: Hereditary Motor Sensory Neuropathy X (HMSN X), 1A, 1B, 4D, 6; mitochondrial; sarcoid
- •
Cerebellum: Friedreich ataxia (FA); Ataxia Telangectasia (AT); Metachromatic Leukodystrophy (MLD); refsum; A-β-lipoproteinemia; Spinocerebellar Ataxia (SCA) 2, 3, 4; Infantile Onset Spinocerebellar Ataxia (IOSCA); Hu & CV-2 autoantibodies
- •
Supratentorial: mitochondrial; thyroid; Hu; B12; vasculitis; neoplastic; sarcoid
- •
Infection: Lyme; HIV; rabies; syphilis; West Nile
- •
Hereditary: polyglucosan; Fabry; HexA; porphyria; prion; ALS; Cowchock; Nicotinamide adenine dinucleotide (NAD); Krabbe; MLD
Face
- •
Bell palsy; Melkersson; Tangier
- •
Polyradiculopathies: sarcoid; Lyme; Guillain-Barré syndrome
- •
Motor neuron disorders: ALS; Kennedy; Möbius
- •
Rule out: MG; myopathy
A history should be obtained regarding dark-colored urine or hematuria as a clue regarding rhabdomyolysis. Myoglobinuria may be associated with: glycogenolysis; CPT II; LPIN1; malignant hyperthermia; central core disease; King-Denborough; DMD (some); hypokalemia; licorice; Li; thiazide; amphotericin; laxatives; infections; mitochondrial myopathy; muscle trauma; ischemia; overactivity; polymyositis, neuroleptic malignant syndrome; drugs (heroin, phencyclidine, ϵ-aminocaproic acid, clofibrate + renal failure, cyclosporine A+ lovastatin); toxins (eg, venoms; intravenous drugs; oral drugs [Haff]; mushrooms; ethanol).
A thorough anesthetic history should be obtained, as malignant hyperthermia is associated with one of the many subtypes of primary familial malignant hyperthermia (hypokalemic periodic paralysis, or one of the malignant hyperthermia susceptibility (MHS) loci, including MHS1: ryanodine receptor, 19q13 [allelic with central core congenital myopathy]; MHS2: Na + channel [SCNA4], 17q11; MHS3: Ca 2+ channel [CACNL2A], 7q21; MHS4: 3q13; MHS5: Ca 2+ channel [CACNA1S], 1q32; MHS6: 5p; CPT2: 1p32) and King-Denborough syndrome. Other NMD conditions occasionally reported to be associated with malignant hyperthermia include DMD, BMD, Fukuyama congenital muscular dystrophy, LGMD, facioscapulohumeral muscular dystrophy (FSHD), periodic paralysis, myotonia congenita, mitochondrial myopathy, minimal change myopathy, myoadenylate deaminase deficiency, and Schwartz-Jampel syndrome.
Neuromuscular disease history
Important elements of NMD history taking are shown in Box 1 .
- •
Distribution of Weakness
- ○
Anatomic distribution/pattern of weakness and focal wasting or hypertrophy of muscle groups (arms versus legs, proximal versus distal, symmetric versus asymmetric)
- ○
Myopathies have weakness that is usually proximal greater than distal with rare exceptions
- ○
- •
Course of weakness
- ○
Acute onset (days to weeks)
- ○
Chronic (months to years)
- ○
Episodic
- ○
Is the weakness getting worse, staying the same, or getting better?
- ○
Ascertain the rate of progression (days, weeks, months, or years)
- ○
- •
Fatigue or lack of endurance
- •
Muscle cramps or stiffness
- •
Lack of sensory loss
- •
Gait characteristics
- ○
Toe walking, excessive lordosis, Trendelenburg or gluteus maximus lurch, and so forth
- ○
- •
Functional difficulties
- ○
Ambulatory distances
- ○
Frequency of falls
- ○
Transitions from the floor to standing
- ○
Problems climbing stairs
- ○
Problems dressing
- ○
Problems reaching overhead
- ○
Difficulty lifting
- ○
Running ability, problems in physical education, and recreational or athletic performance
- ○
- •
Onset age (neonatal, childhood, teen, adult [20–60 years], or geriatric)
- •
Identify factors that worsen or help primary symptoms
- •
History of recent illnesses (eg, recent viral illnesses, respiratory difficulties, pneumonia, pulmonary infections)
- •
Pain
- •
Feeding difficulties, dysphagia, nutritional status, and body composition
- •
Cardiac symptoms (dizziness, syncope, chest pain, orthopnea, cardiac complaints with exertion)
- •
Pulmonary symptoms (breathing difficulties, sleep disturbance, morning headaches)
- •
Anesthetic history (eg, malignant hyperthermia)
- •
History regarding the child’s acquisition of developmental milestones
- ○
Ascertain when the child was able to control his or her head, sit independently, crawl, stand with and without support, walk with and without support, gain fine motor prehension, and acquire bimanual skills (bringing objects to midline, transfer of objects)
- ○
History regarding language acquisition, mental development and school performance
- ○
- •
History regarding pregnancy and neonatal period
- •
Quality of fetal movement, pregnancy complications, perinatal complications, evidence of fetal distress, respiratory difficulties in the recovery room, need for resuscitation or ventilation problems in early infancy, infantile hypotonia, weak cry, poor feeding
The common presenting chief complaints from parents of children with suspected neuromuscular disorders may include infantile floppiness or hypotonia, delay in motor milestones, feeding and respiratory difficulties, abnormal gait characteristics, frequent falls, difficulty ascending stairs or arising from the floor, muscle cramps, or stiffness. Adults often present with chief complaints of strength loss, fatigue or decreasing endurance, falls, difficulty ascending stairs, exercise intolerance, episodic weakness, muscle cramps, focal wasting of muscle groups, breathing difficulties, or bulbar symptoms relating to speech and swallowing.
Respiratory failure due to NMD has been reported in myasthenia gravis, myosin-loss myopathy, acid maltase deficiency, amyloidosis, desmin polymyositis (Jo-1), congenital myopathy (eg, rod; centronuclear), hydroxychloroquine toxicity, neural injury (specifically phrenic lesions), ALS, DMD, and SMA. NMDs with associated cardiac disorders include DMD, BMD, LGMD 1B, LGMD 1C, sarcoglycanopathies, MMD; McLeod, Emery-Dreifuss, Barth syndromes; desmin deficiency; Polymyositis; nemaline rod myopathy; Acid maltase deficiency; debrancher enzyme deficiency; carnitine deficiency; some mitochondrial myopathies; amyloid deficiency; drug-related disorders (metronidazole, emetine, and chloroquine, clofibrate, colchicine); cardiomyopathies; and some periodic paralyses.
Information should be obtained regarding the recent course of the chief complaint, specifically whether the process is getting worse, staying the same, or getting better. If strength is deteriorating, it is important to ascertain the rate of progression (ie, is weakness increasing over days, weeks, months, or years?). It is critical to determine whether the distribution of weakness is predominantly proximal, distal, or generalized. It is also useful to identify factors that may worsen or help the primary symptoms. A history of twitching of muscles may reflect fasciculations. Tremor or balance problems may be due to distal weakness or superimposed cerebellar involvement.
Bulbar involvement may be identified if the individual has difficulty chewing, swallowing, or articulating speech. Visual complaints (blurriness or diplopia) may indicate presence of cataracts or possibly involvement of extraocular musculature. Distal stocking glove or focal sensory complaints may be consistent with a peripheral neuropathy or focal nerve entrapment.
A comprehensive medical history and surgical history should be obtained. A history of recent illnesses should be carefully elucidated, including respiratory difficulties, aspiration pneumonias, or recurrent pulmonary infections. In addition, cardiac symptoms, such as dizziness, syncope, chest pain, orthopnea, or exertional cardiac complaints may indicate superimposed involvement of the myocardium. A pulmonary review of symptoms should be obtained. A history of weight loss may be due to recurrent illnesses, nutritional compromise, swallowing difficulty, or progressive lean tissue atrophy.
For the pediatric patient, a detailed history regarding pregnancy (eg, quality of fetal movement or pregnancy complications) and perinatal problems (evidence of fetal distress, respiratory difficulties in the delivery room, need for resuscitation or ventilation problems in early infancy, ongoing respiratory difficulties, swallowing/feeding difficulties, and persistent hypotonia) should be obtained. Perinatal respiratory distress in the delivery room may be seen in acute infantile type I SMA, myotubular myopathy, congenital hypomyelinating neuropathy, congenital infantile myasthenia, transitory neonatal myasthenia, and severe neurogenic arthrogryposis.
In children, history regarding the acquisition of developmental milestones should be ascertained relating to head control, independent sitting, crawling, standing with and without support, walking with and without support, fine prehension, bimanual skill acquisition (bringing objects to midline, transfer of objects), and language acquisition. Information regarding gait characteristics (toe walking, excessive lordosis, and so forth), running ability, transitions from floor to standing, stair climbing, falls, recreational/athletic performance, pain or muscle cramps, and easy fatigue or lack of endurance may be important clues to the presence of a neuromuscular disorder. History regarding mental development, type of school, and school performance may be important indicators of superimposed central nervous system (CNS) involvement.
For the adult, detailed history regarding the age of onset of symptoms, age when bracing was provided to maintain ambulation, age to use of wheelchair (if applicable), pattern of progression, distribution of weakness, presence of muscle cramps, fatigue, or episodic weakness, presence of atrophy or fasciculations, performance in physical education, military or vocational performance and pursuits, current and past ambulatory distances, ability to transition from floor to standing, problems climbing stairs, and problems reaching overhead or dressing may all be important functional information.
Potential causes of muscle cramps are shown in Box 2 . Muscle cramps in the setting of an elevated creatine kinase (CK) value and no skeletal muscle weakness has been reported, and a pedigree with mild Becker muscular dystrophy. The etiology of myalgias can be quite varied, and a definitive cause is found in only one-fourth of those patients presenting with muscle pain as a chief complaint. Patterns of weakness in myopathies, NMJ disorders, anterior horn cell disorders, and diagnostic considerations are outlined in Box 3 , and selective anatomic distribution of peripheral neuropathies and neuronopathies are listed in Box 4 .
- 1.
Cramps at rest (usually not a neuromuscular disorder)
- a.
Benign nocturnal leg cramps
- b.
Diurnal cramps related to exercise
- a.
- 2.
Cramps occurring with exertion, relieved by rest (may be associated with myoglobinuria)
- a.
Muscular dystrophy, Duchenne, Becker, limb-girdle muscular dystrophy (LGMD)
- b.
Myopathy: rippling muscle syndromes (RMD)
- i.
RMD1: chromosome 1q41; dominant
- ii.
RMD2: caveolin-3; chromosome 3p25.3; dominant
- i.
- c.
Metabolic disorders
- i.
Glycogenoses
- 1.
Myophosphorylase deficiency (type V; McArdle disease)
- 2.
Phosphofructokinase deficiency (type VII)
- 3.
Phosphorylase b kinase deficiency (type VIII)
- 4.
Phosphoglycerate kinase deficiency (type IX)
- 5.
Phosphoglycerate mutase deficiency (type X)
- 6.
Lactate dehydrogenase deficiency (type XI)
- 7.
Myoadenylate deaminase deficiency
- 1.
- ii.
Lipid metabolism disorders
- 1.
Carnitine palmityl transferase deficiency
- 1.
- iii.
Uremia
- iv.
Electrolyte abnormality: hyponatremia, hypocalcemia, hypomagnesemia, hypoglycemia
- v.
Hypothyroidism
- vi.
Hypoadrenalism
- vii.
Paroxysmal myoglobinuria
- viii.
Idiopathic rhabdomyolysis
- i.
- d.
Toxic myoglobinuria
- i.
Alcohol
- ii.
Barbiturates
- iii.
Heroin
- iv.
Carbon monoxide
- v.
Amphotericin B
- vi.
Toxic venoms
- i.
- e.
Inflammatory myositis
- i.
Acute dermatomyositis, polymyositis
- ii.
Viral myositis (Coxsackie and so forth)
- iii.
Bacterial myositis (staphylococci, clostridia)
- i.
- f.
Acute extracellular volume depletion
- i.
Perspiration
- ii.
Diarrhea, vomiting
- iii.
Diuretic therapy
- iv.
Hemodialysis
- i.
- g.
Other lower motor neuron disorders
- i.
Amyotrophic lateral sclerosis (ALS), old polio, other motor neuron disorders
- ii.
Radiculopathy and neuropathy
- i.
- a.
Extraocular Muscles (EOM) Weak
- •
Myasthenia gravis (MG)
- •
Thyroid
- •
Botulism
- •
Mitochondrial: Kearns-Sayre; progressive external ophthalmoplegia (PEO); MNGIE
- •
Myopathy: centronuclear; multicore
- •
Oculopharyngeal muscular dystrophy
- •
Inclusion body myositis (IBM) + contracture
- •
Oculopharyngodistal myopathy
- •
Congenital ophthalmoplegias
Periocular Without EOM Weakness
- •
Dystrophies: myotonic; facioscapulohumeral muscular dystrophy (FSHD); oculopharyngeal
- •
NMJ: MG
- •
Congenital myasthenic syndromes
- •
Congenital myopathies
- •
Inflammatory myopathy: polymyositis
- •
Rule out: VII nerve lesion
Bulbar Dysfunction
- •
NMJ: MG; congenital myasthenic syndromes
- •
Thyroid
- •
Cranial nerve Δ
- •
Oculopharyngeal muscular dystrophy
- •
Distal myopathy (MPD2)
- •
Polymyositis: IBM; scleroderma
- •
Motor neuron Δ: ALS; bulbospinal muscular atrophy (BSMA)
- •
Pseudobulbar; Fazio-Londe
- •
Brown-Vialetto-van Laere
Posterior Neck Weak
- •
Common: MG; polymyositis; ALS
- •
Focal myopathy: neck; paraspinous
- •
Rare: FSHD; LMN (lower motor neuron) syndrome; IBM rod; proximal myotonic myopathy (PROMM); acid maltase deficiency; hypo K + ; carnitine deficiency; endocrine deficiency; desmin deficiency
Proximal Arms Weak
- •
Dystrophy: scapuloperoneal; FSHD
- •
Inflammatory: brachiocervical inflammatory myositis (BCIM)
- •
Absent muscles; Shoulder joint Δ
- •
NMJ: MG
- •
Neuropathic: ALS; pure LMN
- •
Brachial plexopathy
Distal and Proximal Weakness
- •
Dystrophy: myotonic; FSH, scapuloperoneal
- •
Myopathy: congenital; distal
- •
Glygogenoses: debrancher
- •
Phosphorylase b kinase
- •
Neuropathy + Myopathy: paraneoplastic; sarcoid; mitochondria; human immunodeficiency virus (HIV)
- •
Drugs (amiodarone; doxorubicin; colchicine; chloroquine)
Acute Weakness
- •
NMJ: MG
- •
Myoglobinuria
- •
Myosin-loss myopathy
- •
Carnitine deficiency
- •
Periodic paralysis: X-episodic Xp22
- •
Hypo K + : CACNA1S; SCN4A; KCNE3
- •
Hyper K + : SCN4A; KCNE3
- •
Andersen: KCNJ2
- •
Electrolyte disorders: Hyperkalemia, hypokalemia, hypermagnesemia or hypophosphatemia
- •
Barium
- •
Rule out: Neuropathy (acute inflammatory demyelinating polyradiculoneuropathy [AIDP]), chronic inflammatory demyelinating polyradiculoneuropathy [CIDP]); spinal cord
Wasting > Weakness
- •
Pathology: type II atrophy
- •
Cachexia: weight loss >15%, aging/sarcopenia
- •
Disuse
- •
Steroid myopathy
- •
Paraneoplastic
Weakness > Wasting
- •
Polymyositis
- •
Myoglobinuria
- •
Periodic paralysis
- •
NMJ: MG; Congenital myasthenia
- •
Neuropathy + conduction block
Quadriceps Weak
- •
LGMD: 1B; 2B; 2H; ring fiber
- •
Becker
- •
Myositis: IBM; mitochondria; focal
- •
Nerve: femoral; lumbosacral plexopathy
- •
Diabetic amyotrophy; L3-L4 root
Extraocular Muscle
- •
Botulism
- •
Diabetes
- •
Miller-Fisher
- •
Diphtheria
- •
Rule out: MG; myopathy
Proximal Motor
- •
Immune demyelinating: Guillain-Barré syndrome; CIDP; SMA; porphyria
- •
Plexopathy: brachial; lumbar
- •
Rule out: joint pain; myopathy
Proximal Sensory
- •
Hereditary: porphyria; Tangier
- •
Neuronopathy: Hu; Sjögren; thoracic neuropathy
- •
Rule out: myelopathy
Skin Temperature–related
- •
Leprosy
- •
Upper extremity
- •
Immune: multifocal motor neuropathy (MMN); vasculitis; CIDP variant
- •
Amyloid: Carpal tunnel
- •
Entrapment: hereditary neuropathy with liability to pressure palsies (HNPP); other
- •
Toxic: lead; vincristine; ALS; LMN
- •
Rule out: spinal; CNS
Asymmetric
- •
Mononeuritis multiplex
- •
Neuronopathy: ALS; sensory
- •
Entrapments
- •
Plexopathies
- •
Toxic
Mononeuritis Multiplex
- •
Vasculopathy
- •
Amyloid
- •
Leprosy
- •
Diabetes
- •
Cytomegalovirus
- •
Waldenström
- •
Perineuritis
- •
Demyelinating: HNPP; multifocal CIDP; MMN
- •
Compression: multiple
- •
Lymphoma: intraneural
- •
Wartenberg
CNS
- •
Spinal: organophosphate; hexacarbon; Adrenomyeloneuropathy (AMN); metachromatic leukodystrophy (MLD); lymphoma; Cuban; Vernant
- •
Optic: disulfiram; CS2; Hg; drugs; Neuropathy, ataxia, and retinitis pigmentosa (NARP); Charcot Marie Tooth 6 (CMT6); Post col & Retinitis pigmentosa (RP); Cuban; Vernant
- •
Hearing loss: Hereditary Motor Sensory Neuropathy X (HMSN X), 1A, 1B, 4D, 6; mitochondrial; sarcoid
- •
Cerebellum: Friedreich ataxia (FA); Ataxia Telangectasia (AT); Metachromatic Leukodystrophy (MLD); refsum; A-β-lipoproteinemia; Spinocerebellar Ataxia (SCA) 2, 3, 4; Infantile Onset Spinocerebellar Ataxia (IOSCA); Hu & CV-2 autoantibodies
- •
Supratentorial: mitochondrial; thyroid; Hu; B12; vasculitis; neoplastic; sarcoid
- •
Infection: Lyme; HIV; rabies; syphilis; West Nile
- •
Hereditary: polyglucosan; Fabry; HexA; porphyria; prion; ALS; Cowchock; Nicotinamide adenine dinucleotide (NAD); Krabbe; MLD
Face
- •
Bell palsy; Melkersson; Tangier
- •
Polyradiculopathies: sarcoid; Lyme; Guillain-Barré syndrome
- •
Motor neuron disorders: ALS; Kennedy; Möbius
- •
Rule out: MG; myopathy
A history should be obtained regarding dark-colored urine or hematuria as a clue regarding rhabdomyolysis. Myoglobinuria may be associated with: glycogenolysis; CPT II; LPIN1; malignant hyperthermia; central core disease; King-Denborough; DMD (some); hypokalemia; licorice; Li; thiazide; amphotericin; laxatives; infections; mitochondrial myopathy; muscle trauma; ischemia; overactivity; polymyositis, neuroleptic malignant syndrome; drugs (heroin, phencyclidine, ϵ-aminocaproic acid, clofibrate + renal failure, cyclosporine A+ lovastatin); toxins (eg, venoms; intravenous drugs; oral drugs [Haff]; mushrooms; ethanol).
A thorough anesthetic history should be obtained, as malignant hyperthermia is associated with one of the many subtypes of primary familial malignant hyperthermia (hypokalemic periodic paralysis, or one of the malignant hyperthermia susceptibility (MHS) loci, including MHS1: ryanodine receptor, 19q13 [allelic with central core congenital myopathy]; MHS2: Na + channel [SCNA4], 17q11; MHS3: Ca 2+ channel [CACNL2A], 7q21; MHS4: 3q13; MHS5: Ca 2+ channel [CACNA1S], 1q32; MHS6: 5p; CPT2: 1p32) and King-Denborough syndrome. Other NMD conditions occasionally reported to be associated with malignant hyperthermia include DMD, BMD, Fukuyama congenital muscular dystrophy, LGMD, facioscapulohumeral muscular dystrophy (FSHD), periodic paralysis, myotonia congenita, mitochondrial myopathy, minimal change myopathy, myoadenylate deaminase deficiency, and Schwartz-Jampel syndrome.
Family history
Whenever a neuromuscular disorder is suspected with a potential genetic origin, a detailed family history and pedigree chart is absolutely essential. Autosomal dominant conditions may have pedigrees with multiple generations affected with equal predilection to males and females. Typically one-half of offspring within a pedigree are affected. In autosomal recessive conditions, only one generation may be affected with equal proportions of males and females. Proportionally, one-fourth of offspring are clinically affected. Parents in earlier generations may be unaffected and the parents of affected children are presumptive heterozygote carriers of the condition. In many instances of autosomal recessive inheritance, no other family members within the nuclear family unit are affected, making the confirmation of inheritance pattern difficult without a molecular genetic marker being present or protein abnormality confirmed by immunohistochemistry techniques. In X-linked recessive conditions, males on the maternal side of the family are affected in approximately 50% of instances and females are carriers in 50% of instances.
Often it is valuable to examine affected relatives who may be either earlier or later in the course of their NMD relative to the patient. In addition, medical records and diagnostic evaluations of affected family members should be reviewed and the diagnosis confirmed if possible.
In some instances, the examination of a parent can help establish the diagnosis in an affected infant or child, as is frequently the case in MMD. In this disorder, genetic anticipation with abnormal CTG trinucleotide expansion of unstable DNA results in progressively earlier onset of the disease in successive generations with increasing severity, as described elsewhere in this issue.
In the case of dystrophic myopathies, a definitive molecular genetic or pathologic diagnosis established in a sibling or close relative may allow the clinician to establish the diagnosis in a child or adult based on clinical examination and laboratory data such as CK or molecular genetic testing, thus allowing the avoidance of further invasive testing such as a muscle biopsy.
Physical examination
Inspection at Rest
Simple inspection allows the observation of focal or diffuse muscle wasting, or focal enlargement of muscles as with the “pseudohypertrophy” seen in dystrophic myopathies such as DMD and BMD ( Fig. 1 ), LGMD, and lipodystrophy. Cros and colleagues have demonstrated that the increase in calf circumference in DMD is caused by an increase in fat and connective tissue, and is not secondary to true muscle fiber hypertrophy in the gastrocnemius. By contrast, the reduced bulk of the quadriceps in DMD was caused by more severe fiber loss in a more active dystrophic process affecting the knee extensors. In DMD, pseudohypertrophy may be present in other muscle groups such as the deltoid ( Fig. 2 ).
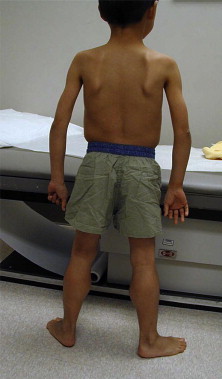
Other neuromuscular disorders may show calf pseudohypertrophy. Calf hypertrophy is particularly prominent in childhood type of acid maltase deficiency. In SMA type III (Kugelberg-Welander syndrome), calf enlargement has been occasionally noted but wasting of affected musculature is typically more prominent. Other NMDs with enlarged muscles include myotonia conditions with overusage; hypothyroidism, acromegaly, infection with cysticercosis, trichinosis and schistosomiasis, anabolic drugs (eg, β2-adrenergic; androgen), glycogen storage diseases, amyloidosis, accumulation of gangliosides, and Schwartz-Jampel syndrome.
Children aged 6 to 11 years with DMD have been noted to exhibit an unusual clinical examination sign caused by selective hypertrophy and wasting in different muscles of the same region. When viewing these patients posteriorly with their arms abducted to 90° and elbows flexed to 90°, the DMD patients demonstrated a linear or oval depression (due to wasting) of the posterior axillary fold with hypertrophied or preserved muscles on its 2 borders (ie, infraspinatus inferomedially and deltoid superolaterally), as if there was a valley between the 2 mounts, as seen in Fig. 2 .
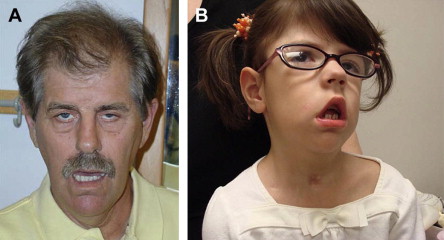
There are several characteristic facial features of MMD that may be noted on inspection ( Fig. 3 ). The adult with long-standing MMD often has facial features so characteristic that it is often easy to make a tentative diagnosis from across the room. The long thin face with temporal and masseter wasting is drawn and described by some as lugubrious. Adult males often exhibit frontal balding. Infants and young children with a variety of myopathies may exhibit a tent-shaped mouth (see Fig. 3 ).
Focal atrophy of particular muscle groups may provide diagnostic clues to specific neuromuscular disorders. SMA shows diffuse muscle atrophy or focal atrophy in more slowly progressive subtypes. Emery-Dreifuss may present with striking wasting of the biceps, accentuated by sparing of the deltoids and forearm muscles. There may also be wasting of the calf muscles in this condition. Quadriceps-selective weakness and atrophy may be a presenting sign in a variety of myopathies such as BMD, LGMD 1B; 2B; 2H; 2L (11p13 LGMD 2L: recessive, anoctamin 5 [ANO5, MEM16E, GDD1]; chromosome 11p14.3; recessive, Emery-Dreifuss: lamin A/C hereditary IBM3), inflammatory myopathies, sporadic inclusion body myositis (IBM), polymyositis with mitochondrial pathology, focal myositis, myopathy with ringed fibers, and SMA types III and IV, 5q types III and IV, femoral neuropathy, diabetic amyotrophy, and L3-L4 radiculopathy.
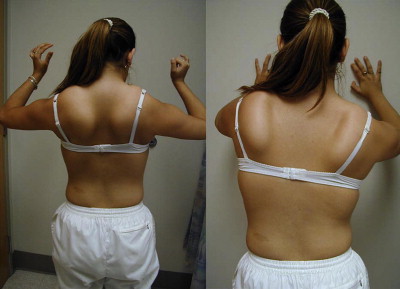
Patients with focal shoulder girdle weakness, as in FSHD and LGMD, may show characteristic patterns of muscle atrophy and scapular displacement. In FSHD, involvement of the latissimus dorsi, lower trapezius, rhomboids, and serratus anterior results in a characteristic appearance of the shoulders with the scapula positioned more laterally and superiorly, giving the shoulders a forward-sloped appearance. The upper border of the scapula rises into the trapezius, giving it a hypertrophied appearance falsely. From the posterior view, the medial border of the scapula may exhibit profound posterior and lateral winging ( Fig. 4 ). The involvement of shoulder girdle musculature in FSHD may also be quite asymmetric.
Most weakness in neuromuscular disorders is associated with focal atrophy. Those with CMT, particularly those with the type II axonal forms, demonstrate distal atrophy or “stork-leg appearance” relatively early in the disease course. Those with primarily demyelinating type I forms of CMT may show distal wasting later in the disease course.
Muscle fasciculations may be seen as nonspecific findings of a variety of lower motor neuron disorders. Fasciculations are particularly common in motor neuron disorders, such as ALS and SMA. Distal essential tremor may be seen in a large proportion of CMT patients (30%–50%), and other patients with weakness such as SMA. Polyminimyoclonus, another variant of muscle fasciculations, characterized by a fine tremor of the fingers and hands, may be evident in SMA I and II.
Palpable nerves in the cubital tunnel, posterior auricular region, or around the fibular head may indicate onion bulbs seen in CMT I subtypes, or Dejerine-Sottas disease (CMT III).
General Examination
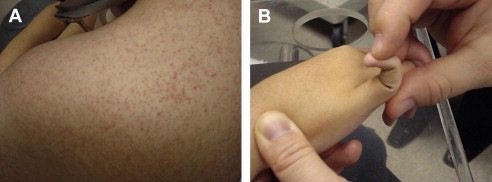
Important aspects of the cardiac and pulmonary assessment pertaining to NMD conditions are described in the next issue of this journal. Hepatomegaly may be seen in metabolic myopathies such as acid maltase deficiency (type 2 glycogenosis) and types 3 and 4 glycogenosis. Characteristic rashes and nail-bed capillary changes may be present in dermatomyositis. Patients with Ullrich congenital muscular dystrophy who have a collagen VI abnormality often show hyperkeratosis pilaris in the extensor surfaces of the upper arms ( Fig. 5 ). Craniofacial changes and dental malocclusion are commonly seen in congenital MMD, congenital myopathies, congenital muscular dystrophy, and type II SMA.
Cognitive Assessment
Some NMDs such as congenital and noncongenital MMD (DM1), PROMM (DM2), Fukuyama congenital muscular dystrophy, selected cases with mitochondrial encephalomyelopathies, and a small proportion of DMD cases may have significant intellectual impairment. In addition, other NMDs with significant cognitive involvement include hereditary IBM (9pp13), selected mitochondrial encephalomyopathies, congenital muscular dystrophy (Santavuori, POMGnT1 1p32; merosin 6q22; Fukuyama fukutin 9q31; and integrin-α7 12q13), and phosphoglycerate kinase deficiency. In these instances referral for neuropsychological testing, a neurodevelopmental evaluation, and/or a pyschoeducational evaluation may be helpful.
Cranial Nerve Examination
Neuromuscular disorders tend not to have optic nerve involvement; however, an evaluation of vision and a fundoscopic examination can be exceedingly important. For example, MMD patients (DM1) may have cataracts giving significant visual impairment. These cataracts may have multicolored subcapsular opacities noted on a careful slit-lamp examination. In addition to the lens opacities, retinal degeneration characterized by peripheral pigmentary changes in the macula may be present in MMD. Other ocular abnormalities, including low intraocular pressure, enophthalmos, blepharitis, and corneal lesions have been described in this disorder. All MMD patients should have regular ophthalmologic evaluations.
Ptosis is a finding described in myasthenia gravis, congenital myasthenic syndromes, transient autoimmune neonatal myasthenia, oculopharyngeal muscular dystrophy, and occasionally MMD.
Ophthalmoparesis may be a finding seen in myasthenia gravis, congenital myasthenic syndromes, and oculopharyngeal muscular dystrophy. In addition, extraocular muscle involvement may occur in some of the congenital myopathies, particularly myotubular myopathy, and some of the mitochondrial myopathies. For example, progressive external ophthalmoplegia (PEO) is a mitochondrial disorder that may present with bilateral ophthalmoplegia with or without limb weakness. Congenital fibrosis of the extraocular muscles, or congenital familial external ophthalmoplegia, is an autosomal dominant, congenital, nonprogressive disorder of the ocular muscles with primary findings of bilateral ptosis and external ophthalmoplegia. Affected individuals with PEO often have associated facial weakness. Gaze is limited in all directions, eye movement speed is slow. the disorder is associated with ptosis and is slowly progressive.
Facial weakness is an important clinical feature of FSHD. The initial weakness affects the facial muscles, especially the orbicularis oculi, zygomaticus, and orbicularis oris. These patients often have difficulty with eye closure but not ptosis ( Fig. 6 ). The individual may assume an expressionless appearance and exhibit difficulty whistling, pursing the lips or drinking through a straw, or smiling. Even in the very early stages, forced closure of the eyelids can be easily overcome by the examiner. Masseter, temporalis, extraocular, and pharyngeal muscles are characteristically spared in FSHD.

Facial weakness may also be observed in oculopharyngeal muscular dystrophy, myasthenia gravis, congenital myasthenic syndromes, Möbius syndrome, congenital myopathies, and myotubular myopathy. Rare cases with FSHD have been described, secondary to a hereditary neuropathy with weakness in predominantly a scapuloperoneal distribution, with involvement of the facial muscles and other limb muscles such as the shoulder girdle, ankle dorsiflexors, and ankle everters.
A sensorineural hearing deficit was originally observed in Coats syndrome (early-onset FSHD). These individuals have a myopathy presenting in infancy. The disease progression is fairly rapid, with most individuals becoming wheelchair reliant by the late second or third decade; they may also have a progressive exudative telangiectasia of the retina. Early recognition and photocoagulation of the abnormal retinal vessels may prevent visual loss. Several studies of later-onset FSHD using audiometry have demonstrated hearing deficits in many patients in addition to those with Coats syndrome, suggesting that impaired hearing function is more common than expected in FSHD. Thus, all patients with FSHD should have screening audiometry and ophthalmologic evaluation.
Involvement of palatal, pharyngeal, and laryngeal muscles may produce dysarthria and dysphagia. Patients at particular risk include those with ALS, SMA, myasthenia gravis, congenital myasthenic syndromes, and congenital myopathies such as myotubular myopathy, oculopharyngeal muscular dystrophy, late-stage DMD, and late-stage LGMD with autosomal recessive inheritance. The function of the swallowing mechanism is best evaluated with a fluoroscopic video-dynamic evaluation of swallowing.
Vocal cord paralysis is a relatively uncommon finding in hereditary neuromuscular disorders; however, distal infantile spinal muscular atrophy with diaphragm paralysis (DSMA1; SMARD1; HMN 6) is linked to chromosome 11q13.3. Vocal cord paralysis has also been described as a complication of dermatomyositis.
Examination of the tongue for muscle bulk and presence of fasciculations should be performed. Tongue fasciculations are a common finding in ALS and SMA types I, II, and III. However, tongue fasciculations are not an absolute finding in ALS or SMA. For example, 56% to 61% of SMA I patients, 30% to 70% of SMA II, patients and roughly half of SMA III patients late in the disease course show tongue fasciculations. Thus, absence of tongue fasciculations does not necessarily exclude these motor neuron disorders. The bulk of the tongue may be increased in some metabolic diseases such as acid maltase deficiency, and often in later stages of DMD.
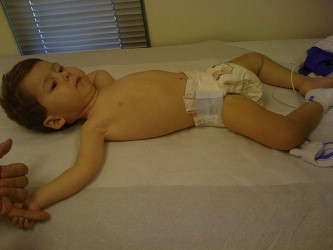
Tone
Hypotonia ( Fig. 7 ) is an important clinical examination finding in children with neuromuscular disorders. The most common etiology for infantile hypotonia is central, accounting for approximately 80% of cases. Hypotonia remains the most common reason for referral to the pediatric electrodiagnostic laboratory. A differential diagnosis of infantile hypotonia is shown in Box 5 .
- 1.
Cerebral hypotonia
- a.
Chromosome disorders
- i.
Trisomy
- ii.
Prader-Willi syndrome
- i.
- b.
Static encephalopathy
- i.
Cerebral malformation
- ii.
Perinatal CNS insult
- iii.
Postnatal CNS insult
- i.
- c.
Peroxisomal disorders
- i.
Cerebrohepatorenal syndrome (Zellweger)
- ii.
Neonatal adrenoleukodystrophy
- i.
- d.
Inborn errors of metabolism
- i.
Glycogen storage disease type II (Pompe disease)
- ii.
Infantile GM1, gangliosidosis
- iii.
Tay-Sachs infantile GM2 gangliosidosis)
- iv.
Vitamin dependency disorders (many) and so forth
- i.
- e.
Amino acid and organic acid disorders
- i.
Maple syrup disease
- ii.
Hyperlysinemia
- iii.
Nonketotic hyperglycinemia
- iv.
Propionyl-CoA carboxylase deficiency and so forth
- i.
- f.
Other genetic disorders
- i.
Familial dysautonomia
- ii.
Cohen syndrome
- iii.
Oculocerebrorenal syndrome (Lowe)
- i.
- g.
Benign congenital hypotonia
- a.
- 2.
Spinal cord
- a.
Trauma (obstetric; postnatal)
- i.
Hypotonia early with acute paraplegia
- ii.
Hypertonia
- i.
- b.
Tumor or arteriovenous malformation
- i.
Hypertonia may occur later or with slow growing tumor
- i.
- c.
Anterior horn cell
- i.
SMA type I (Werdnig-Hoffman)
- ii.
SMA type II
- iii.
Poliomyelitis
- iv.
Neurogenic arthrogryposis
- i.
- a.
- 3.
Polyneuropathies
- a.
Congenital hypomyelinating neuropathy
- b.
Chronic inflammatory demyelinating polyneuropathy
- c.
AIDP (Guillain-Barré)
- d.
Hereditary motor-sensory neuropathies (eg, I, III)
- e.
Toxic polyneuropathy
- f.
Leukodystrophies (Krabbe; Nieman-Pick)
- g.
Leigh syndrome
- h.
Giant axonal neuropathy
- i.
Dysmaturation neuropathy
- a.
- 4.
Neuromuscular junction
- a.
Presynaptic
- i.
Infantile botulism
- ii.
Hypermagnesemia: eclampsia
- iii.
Aminoglycoside antibiotics
- iv.
Congenital myasthenia
- v.
Acetylcholine vesicle paucity
- vi.
Decreased quantal release
- i.
- b.
Postsynaptic
- i.
Neonatal (autoimmune)
- ii.
Congenital myasthenia
- iii.
Acetylcholinesterase deficiency
- iv.
Slow changes
- v.
Acetylcholine receptor deficiency
- i.
- a.
- 5.
Myopathies
- a.
Congenital myopathies
- i.
Nemaline rod
- ii.
Central core
- iii.
Myotubular (centronuclear)
- iv.
Congenital fiber type disproportion
- i.
- b.
Congenital myotonic dystrophy
- c.
Congenital muscular dystrophy
- i.
Fukuyama type (CNS involvement)
- ii.
Merosin deficiency (with or without CNS involvement)
- iii.
Atonic-sclerotic type (Ulrich disease)
- iv.
Undifferentiated
- i.
- d.
Inflammatory myopathies
- i.
Infantile polymyositis
- i.
- e.
Metabolic myopathies
- i.
Acid maltase deficiency (type II)
- ii.
Muscle phosphorylase deficiency (type V)
- iii.
Phosphofructokinase deficiency (type VII)
- iv.
Cytochrome c oxidase
- v.
Carnitine deficiency
- i.
- f.
Endocrine myopathies
- i.
Hypothyroidism
- ii.
Hypoparathyroidism
- i.
- a.
Strength Assessment
The distribution of weakness is often a critical piece of information that allows the clinician to categorize a patient into a specific neuromuscular diagnostic syndrome. The distribution of weakness should be noted (predominantly proximal versus distal; lower extremity versus upper extremity; focal versus generalized; isolated peripheral nerve distribution versus multiple peripheral nerves; or single versus multiple roots/myotomes). It should be noted whether extraocular, facial, and bulbar muscles are involved or spared. In addition to appendicular (limb) strength, the strength of axial musculature should also be noted.
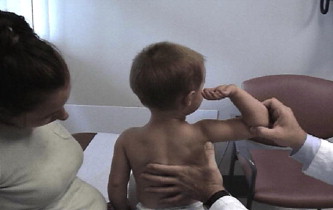
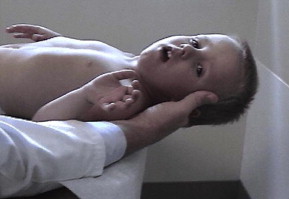
A common finding in myopathies, particularly dystrophic myopathies, is the early and selective weakness of the neck flexors as opposed to the neck extensors. For example, the neck flexors are the earliest muscle group to show weakness in DMD. Clinical examination of a child or adult with a suspected dystrophic myopathy should always include an evaluation of neck-flexor strength ( Fig. 8 ). Quantitative isometric strength measurements of neck strength in normal subjects with grade 5 neck flexors and extensors on manual muscle testing show the neck extensors to be stronger than the neck flexors. Absolute muscle strength is directly proportional to the physiologic cross-sectional area of muscle fiber. The cross-sectional area of the neck extensors is much greater than the cross-sectional area of the neck flexors. Seventeen muscle groups act bilaterally as neck extensors, whereas only 6 muscle groups act bilaterally as neck flexors. Thus, with dystrophic myopathies the progressive loss of muscle fiber over time results in significant clinically detectable weakness of the neck flexors earlier than the neck extensors. This weakness is often accentuated in children by the large proportional size of the head relative to the rest of their body.
Predominantly distal lower extremity weakness is highly suggestive of an acquired or inherited peripheral neuropathy, the differential for which is quite broad. There are several other inherited neuromuscular disorders that can present with distal lower extremity weakness. Anterior horn cell disorders include distal chronic SMA. Myopathies include inflammatory myopathies, such as inclusion body myositis, scapuloperoneal syndromes including scapuloperoneal muscular dystrophy, late adult-onset autosomal dominant distal myopathy, Finnish tibial muscular dystrophy, early adult-onset autosomal recessive distal myopathy (types I and II) and, occasionally, metabolic myopathies. Distal upper extremity weakness may be seen initially in Asian-variant distal SMA, and Welander-type late adult-onset autosomal dominant distal myopathy.
The differential diagnosis of the limb-girdle syndromes presenting in childhood and adulthood, and characterized by predominantly proximal weakness of shoulder and pelvic girdle muscles, remains large and may include LGMD subtypes, polymyositis, dermatomyositis, congenital myasthenic syndromes, IBM, type III SMA, manifesting carrier of DMD, BMD, FSHD, scapuloperoneal myopathy, Emery-Dreifuss muscular dystrophy, congenital myopathies occasionally presenting later in childhood or adulthood (ie, adult-onset nemaline rod disease, central core disease, centronuclear myopathy, fiber type disproportion, multicore disease, sarcotubular myopathy, fingerprint myopathy, reducing body myopathy), mitochondrial myopathies with limb-girdle weakness, other metabolic myopathies that may present in adulthood (ie, adult-onset acid maltase deficiency, debrancher enzyme deficiency, McArdle disease, carnitine deficiency), myopathy with tubular aggregates, and myopathy with cytoplasmic bodies.
Quantitative strength testing
Strength is difficult to objectively evaluate in children with motor impairments. Kilmer and colleagues have demonstrated strength measurement to be more stable and reproducible in children older than 5 years. Quantitative strength measurements have been demonstrated to be far more sensitive than clinical strength testing for detecting weakness in children and adults with motor impairments. The author and his colleagues at the University of California Davis Research and Training Center in Neuromuscular Disease have published several studies using isometric and isokinetic quantitative strength testing as a measure of impaired strength in patients with neuromuscular disorders, and have shown quantitative strength testing to be a more sensitive measure of weakness than clinical examination, particularly when strength is grade 4 to 5 on manual muscle testing. At age 6, the reduction in tension developed by the knee extensors of DMD subjects was approximately 50% of control values for knee extension while knee extension was between grade 4 and 5 on same-day clinical manual muscle testing. Thus, by the time patients have progressed to grade 4 strength by manual muscle testing, substantial weakness is present.
Repetitive strength testing
When suspecting episodic weakness with a fatigue component, the examiner may have the patient repetitively contract a muscle against resistance for 10 to 15 contractions through a functional range of motion. This exercise often brings about obvious fatigue and progressive weakness after several contractions in myasthenic syndromes, such as myasthenia gravis or congenital myasthenia; this can also be accomplished more quantitatively with isokinetic dynamometry, comparing peak torque with initial contractions versus later contractions (eg, the fifth contraction or tenth contraction).
Sensory Examination
A stocking-glove loss of sensation or vague distal dysesthesias may be present in a peripheral neuropathy. Focal sensory changes in one or more peripheral nerve distributions can indicate focal entrapments, which are commonly seen in hereditary neuropathy with predisposition to pressure palsy (HNPP), one of the CMT subtypes.
Cerebellar Examination
The presence of tremor, dysdiadachokinesia (problems with rapid alternating movements), or axial and appendicular ataxia/balance problems can be important findings in syndromes such as ataxia telangiectasia, autosomal dominant spinocerebellar degeneration syndromes, and Friedreich ataxia.
Deep Tendon Reflexes
Whereas deep tendon reflexes (DTRs) are generally depressed or absent in many NMDs, they may be brisk in syndromes with superimposed upper motor neuron involvement such as ALS or some spinocerebellar degeneration syndromes. It is important to remember that the presence of DTRs does not necessarily exclude the presence of an NMD. For example, in one series, DTRs were absent in all 4 extremities in 74% of SMA I cases, but present and depressed in 26% of cases. In SMA II and III, DTRs are invariably depressed and usually become absent over time.
Myotonia
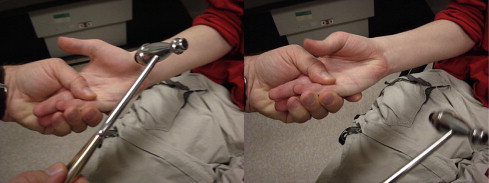
The clinical finding common to all myotonic disorders is myotonia, which is a state of delayed relaxation or sustained contraction of skeletal muscle. Grip myotonia may be demonstrated by delayed opening of the hand with difficult extension of the fingers following tight grip. Paradoxic myotonia is the situation whereby myotonia becomes worse with successive movements instead of improving with activity. Percussion myotonia may be elicited by percussion of the thenar eminence with a reflex hammer, giving an adduction and flexion of the thumb with slow return ( Fig. 9 ). Other sites that may give a local contraction with percussion include the deltoid, brachioradialis, and gluteal muscles. Occasionally, myotonia of the tongue draped over a tongue blade may be elicited with a midline tap of the finger, giving a bilateral contraction notch along the lateral portion of the tongue bilaterally with slow relaxation. Myotonic syndromes include MMD (Steinert disease), myotonia congenita (Thomsen disease), Becker-type myotonia congenita, paramyotonia congenita (Eulenburg disease), and Schwartz-Jampel syndrome (chondrodystrophic myotonia).
Related posts:
 Patient Safety in Cancer Rehabilitation
The Utility of Electromyography and Mechanomyography for Assessing Neuromuscular Function: A Noninvasive Approach
Novel Concepts Integrated in Neuromuscular Assessments for Surgical Restoration of Arm and Hand Function in Tetraplegia
Cardiac MRI in Muscular Dystrophy: An Overview and Future Directions
New Opportunities and Novel Paradigms to Support Neuromuscular Research
Neuromuscular Ultrasonography: Quantifying Muscle and Nerve Measurements
Patient Safety in Cancer Rehabilitation
The Utility of Electromyography and Mechanomyography for Assessing Neuromuscular Function: A Noninvasive Approach
Novel Concepts Integrated in Neuromuscular Assessments for Surgical Restoration of Arm and Hand Function in Tetraplegia
Cardiac MRI in Muscular Dystrophy: An Overview and Future Directions
New Opportunities and Novel Paradigms to Support Neuromuscular Research
Neuromuscular Ultrasonography: Quantifying Muscle and Nerve Measurements
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


