Chondrosarcoma (Primary, Secondary, Dedifferentiated, and Clear Cell)
CHONDROSARCOMA
Chondrosarcoma should be differentiated from osteosarcoma because of basic pathologic differences that are reflected in vastly different clinical, therapeutic, and prognostic features. The exact origin of chondrosarcoma is obscure, but the salient pathologic fact is that its basic proliferating tissue is cartilaginous throughout. Large portions of these tumors may become myxomatous, calcified, or even ossified. Sometimes at the periphery of lobules of high-grade chondrosarcoma, a few fibrosarcoma-like spindling tumor cells occur. Osseous trabeculae, when present, are seen at the periphery of lobules and appear to be rimmed with osteoblasts. However, when the malignant cells directly produce an osteoid lacework or osteoid trabeculae even in small foci, the neoplasm has clinical characteristics of osteosarcoma and belongs in that category (Table 6.1).
Chondrosarcoma usually has a slow clinical evolution. Metastasis is relatively rare and often occurs late. Therefore, the basic therapeutic goal is local control; however, in osteosarcoma, systemic spread has also to be addressed because of early hematogenous dissemination. Attainment of this goal demands adequate, frequently radical, early surgical treatment but generally not chemotherapy.
Chondrosarcomas can arise de novo in extraskeletal tissues or in teratomas and other mixed tumors. Chondrosarcomas composed of hyaline cartilage are extremely rare in somatic soft tissues.
Secondary chondrosarcomas arise most commonly in osteochondromas (osteocartilaginous exostosis), especially in the multiple, familial type. Whether a solitary enchondroma can give rise to a chondrosarcoma is a difficult question to answer.
The distinction between an enchondroma and an extremely well-differentiated chondrosarcoma depends so much on the radiographic findings that a pure histologic interpretation is hazardous. It is possible that enchondromas undergo change to chondrosarcoma, but this is hard to prove one way or the other. In the Mayo Clinic series of 158 definite secondary chondrosarcomas (in 155 patients), 82 occurred in patients with solitary exostoses, 44 in patients with multiple hereditary exostoses, and 19 in patients with multiple chondromas (8 with Ollier disease, 5 with Maffucci syndrome, and 6 with multiple chondromas). The remaining 10 occurred in miscellaneous conditions such as fibrous dysplasia and previous radiation. Some of the details of these secondary chondrosarcomas are given in Chapters 2 and 3.
In addition to the 1,223 primary and secondary chondrosarcomas (in 1,228 patients), 145 had given rise to more highly malignant tumors—osteosarcomas, fibrosarcomas, or malignant fibrous histiocytomas— and these are called dedifferentiated chondrosarcomas.
Among the primary chondrosarcomas were 26 of the clear cell type. The 46 mesenchymal chondrosarcomas, not included in this list, are discussed in Chapter 7.
INCIDENCE
Chondrosarcomas constituted just over 20.4% of the malignant tumors in our series, and approximately 75% were of the primary type. Dedifferentiation has occurred in both primary and secondary chondrosarcomas. Osteosarcoma was approximately 1.5 times as common as chondrosarcoma.
TABLE 6.1. Classification of Chondrosarcoma | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
SEX
Approximately 57% of the patients were males.
AGE
Chondrosarcoma is primarily a tumor of adulthood and old age. Approximately 60% of the patients were in the fourth, fifth, and sixth decades of life. Only seven patients were in the first decade of life. The youngest patient was a 3-year-old boy with a lesion of the mid right humerus, and the oldest was a 91-year-old woman with a lesion of the tenth thoracic vertebra. Patients with secondary chondrosarcoma were somewhat younger, with approximately 52% being in the third and fourth decades of life. Only 5.21% of the total group of patients was in the second decade of life. Any series with a relatively large number of patients in the first two decades of life probably includes patients with chondroblastic osteosarcoma, a tumor with biologic capabilities similar to that of the overall osteosarcoma group.
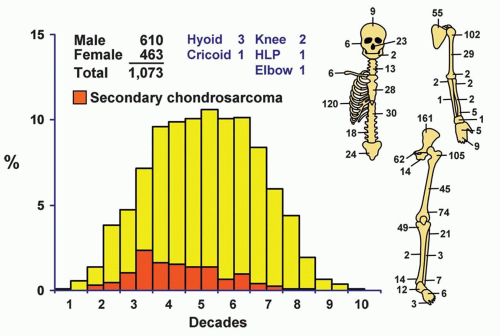 Figure 6.1. Distribution of chondrosarcomas according to age and sex of the patient and site of the lesion. |
LOCALIZATION
More than two-thirds of the tumors were in the trunk (including the shoulder girdle) and the upper ends of the femora and humeri (Fig. 6.1). Most of the lesions in the maxillary region seemed to arise from the cartilage of the walls of the nasal cavity. There were 23 tumors in the nose, whereas only 6 tumors were considered to arise primarily in the maxilla and only 2 in the mandible. Three tumors arose from the hyoid bone; one of the patients also had Gardner syndrome. Five chondrosarcomas involved the synovium. Two of these were considered to be primary chondrosarcoma of the synovium: one in the hip and the other in the knee. Three were considered to be secondary to preexisting synovial chondromatosis: one each involved the knee, ankle, and elbow. In addition, five chondrosarcomas arose after radiation therapy; one of the patients had fibrous dysplasia. One of the postradiation chondrosarcomas was a clear cell variant. Three patients had chondrosarcoma arising in preexisting fibrous dysplasia. One of these patients previously had radiation, and that tumor is included among the five arising after radiation. One of the other two tumors was a clear cell chondrosarcoma. The remarkable rarity of chondrosarcoma in the distal portions of the extremities, with only 36 occurring distal to the ankle and wrist joints, is noteworthy. Some of the few lesions reported as juxtacortical chondrosarcomas in the literature are classed with chondroblastic osteosarcomas because component malignant cells produce osteoid; these are described in a later chapter as periosteal osteosarcomas. However, 24 chondrosarcomas were situated peripherally. Seventeen of these were termed periosteal chondrosarcomas because their radiographic features suggested a relation to periosteal chondroma. The remaining seven were classified as peripheral chondrosarcomas because they did not show
features similar to those of periosteal chondromas and did not appear to have a preexisting osteochondroma.
features similar to those of periosteal chondromas and did not appear to have a preexisting osteochondroma.
Localization data on the 158 chondrosarcomas secondary to exostoses and multiple chondromas are given in Figure 6.2. As indicated previously, the 44 patients with chondrosarcomas complicating multiple exostoses were from a group of 184 patients who required surgery for the latter condition. Of the 966 patients requiring surgery for solitary exostoses, 82 had complicating chondrosarcomas. Nineteen of the 73 patients operated on for multiple chondromas of the skeleton had secondary chondrosarcomas. These data should not be construed to represent the true incidence of sarcomatous change in the three conditions. Continued follow-up of the total groups should alter the data, and factors of selection probably increase the likelihood that the patients with sarcoma will seek help at a large medical center. Figure 6.1 shows that patients with secondary chondrosarcoma tend to be younger than the average patient with primary chondrosarcoma.
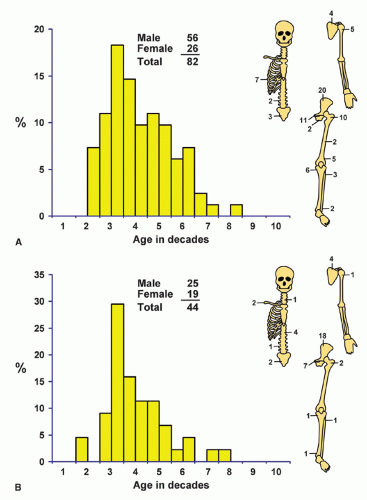 Figure 6.2. Distribution of secondary chondrosarcomas according to age and sex of the patient and site of the lesion. A: Solitary exostosis. B: Multiple exostoses. C: Chondrodysplasias. D: Total. |
Information concerning various subtypes of chondrosarcoma is given in Table 6.1.
SYMPTOMS
Local swelling and pain, either alone or in combination, are significant presenting symptoms. Pain strongly suggests active growth of a central cartilaginous tumor. Except for some tumors of the pelvic girdle or spinal column, in which referred pain may precede local pain or discernible physical or radiographic findings, localization of these tumors is easy. As in other tumors of bone, the characteristics of the pain or swelling offer little aid in the differential
diagnosis. Pathologic fracture may be the presenting symptom. The prolonged clinical course so often observed affords a clue. A tumor that gradually enlarges for one to two decades (or even longer) may have been noted by patients who have an osteochondroma that undergoes malignant change. Such transformation often produces pain and rapid increase in the size of a tumor of long duration. Patients with primary chondrosarcoma also may have had symptoms for several years before seeking definitive therapy. Inadequately treated tumors have a typical history of many recurrences and, finally, of inoperable extension or metastasis leading to death. A few chondrosarcomas have a rapid clinical course because of a higher degree of malignancy initially or because of increased activity with recurrences.
diagnosis. Pathologic fracture may be the presenting symptom. The prolonged clinical course so often observed affords a clue. A tumor that gradually enlarges for one to two decades (or even longer) may have been noted by patients who have an osteochondroma that undergoes malignant change. Such transformation often produces pain and rapid increase in the size of a tumor of long duration. Patients with primary chondrosarcoma also may have had symptoms for several years before seeking definitive therapy. Inadequately treated tumors have a typical history of many recurrences and, finally, of inoperable extension or metastasis leading to death. A few chondrosarcomas have a rapid clinical course because of a higher degree of malignancy initially or because of increased activity with recurrences.
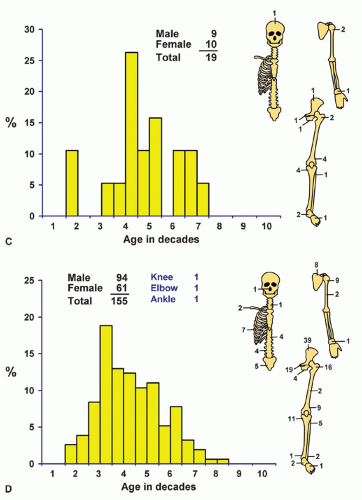 Figure 6.2. Continued. |
The slow clinical evolution of chondrosarcoma is emphasized by the finding that in approximately 10% of chondrosarcomas that recurred in the Mayo Clinic series, the interval between treatment and recurrence was 5 to 10 years. A recurrence may even become manifest more than 10 years after initial treatment. Because recurrence may be so delayed, conclusions about the efficacy of any treatment must be based not only on a sizeable series of cases but also on follow-up of 10 years or more. In a selected series of patients treated at Mayo Clinic, Bjornsson and coauthors found that the recurrence rate was approximately 20%. The cumulative probability of local recurrence was 20.1% at 5 years, 22.4% at 10 years, and 26.5% at 20 years. This study emphasizes the possibility of local recurrence even up to 20 years.
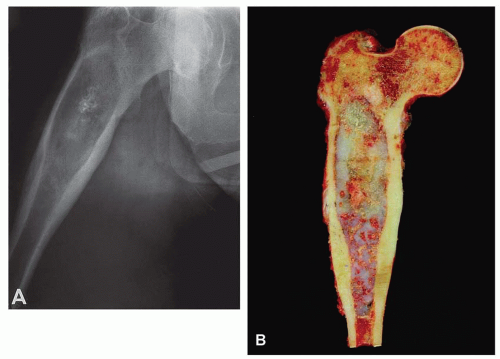 Figure 6.3. Proximal femoral chondrosarcoma in a 77-year-old woman. A: Plain radiograph shows that the lesion is focally mineralized. The combination of cortical expansion and cortical thickening suggests the diagnosis. B: Corresponding gross specimen. There is marked thickening of the cortex, and the tumor erodes the cortex at several places. (Figure 6.3A is from Unni, K. K., and Inwards, C. Y.: Tumours of Osteoarticular System. In: Fletcher, C. D. M. [ed]. Diagnostic Histopathology of Tumors. Edinburgh, Churchill Livingstone, 1995. By permission of the publisher.) |
 Figure 6.4. Chondrosarcoma arising in the proximal femur. The plain radiograph shows a lytic lesion with mineralization, cortical thickening, and endosteal scalloping. |
PHYSICAL FINDINGS
Many chondrosarcomas can be palpated, but a sizeable number of those affecting the trunk, or even the long bones of the extremities if they have not breached the cortex, cause pain alone to signal their presence. A palpable mass is characteristically hard and may be painful. When a mass cannot be palpated, the diagnosis may be difficult, especially for chondrosarcomas of the innominate bone, which may lack definite radiographic changes. The region of the acetabulum, where many chondrosarcomas originate, is notorious for such “hidden” malignant tumors. In our series, one chondrosarcoma of the hyoid bone was associated with Gardner syndrome.
RADIOGRAPHIC FEATURES
The radiograph is nearly always helpful and often affords almost pathognomonic evidence of chondrosarcoma (Figs. 6.3, 6.4, 6.5, 6.6 and 6.7). Chondrosarcomas tend to be large. In a study by Bjornsson and coworkers, the mean size of the tumors of the 180 cases for which radiographs were available was 9.5 cm. Small lesions tended to be round or oval. Large lesions, once they reached the adjacent cortex, had a pronounced tendency to conform to the shape of the bone. In this study, approximately 79% of the tumors had poor margination, 17% had intermediate margins, and 4% were sharply circumscribed. Analysis of margination was difficult in several intramedullary tumors because only matrix calcification was visualized. A clear-cut sclerotic rim was present in only two tumors. Approximately three-fourths of the lesions showed mineral, which was considered to be slight in 39% of tumors, moderate in 47% and marked in 14%. In some lesions, calcification not visible on other studies was clearly demonstrable on computed tomograms. Areas of lucency within an otherwise calcified tumor were common and considered suggestive of chondrosarcoma. Approximately one-fourth of the tumors did not show calcification. However, these were considered to have malignant characteristics on the basis of other features. Approximately 84% of the tumors had caused cortical abnormalities. More than half of these consisted of endosteal erosion, and the rest showed frank cortical destruction. Endosteal scalloping is a sign of growth but not necessarily of chondrosarcoma. Enchondromas of small bones always show erosion of cortex; permeation of the tumor through the cortex into soft tissue has to be identified before a diagnosis of chondrosarcoma is entertained in the small bones. More than one-third of the lesions showed widening or expansion of the bone. Of these, approximately one-half had cortical thinning. Approximately 20% of the tumors showed the combination of expansion of bone and cortical thickening. Periosteal new bone formation was unusual and, when present, scant. Forty percent of the lesions had a soft-tissue mass. Magnetic resonance images and computed tomograms detected a soft-tissue mass and delineated its extent more accurately than conventional radiographs (Figs. 6.8, 6.9 and 6.10).
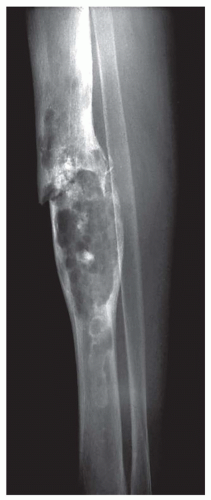 Figure 6.5. Plain radiograph of a chondrosarcoma in the mid-tibia in a 71-year-old woman. The lesion is expansile with focal mineralization and a pathologic fracture (Case provided by Dr. Vandana Kumar, London, Kentucky). |
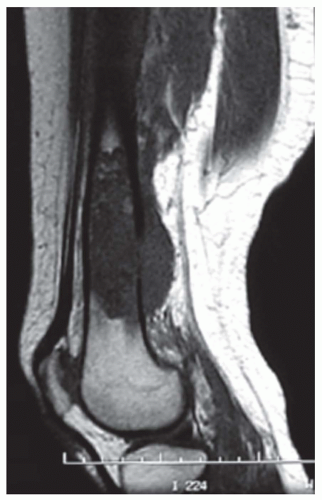 Figure 6.6. Magnetic resonance image of a grade 1 chondrosarcoma involving the distal femur with a soft-tissue mass. This imaging modality can be helpful in evaluating the extent and character of intramedullary disease and soft tissue extension. |
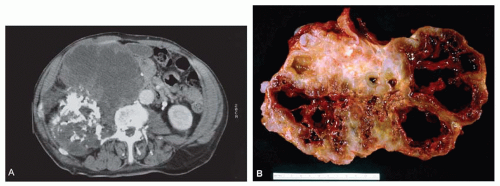 Figure 6.7. Huge chondrosarcoma apparently arising from the vertebral column in a 74-year-old man was manifested by an abdominal mass. A: Focal calcification is apparent on computed tomography. B: Gross specimen of the chondrosarcoma. The tumor shows focal calcification and pronounced cystification because of the myxoid change in the matrix. The size of the lesion and the cystification alone are sufficient to diagnose chondrosarcoma. |
In an osteochondroma that has undergone malignant change, the radiographic findings may be similar to those of the benign lesion from which it originated, but the surface ordinarily is indistinct and fuzzy, and the clear demarcation from the adjacent soft tissue may be lost. A large soft-tissue mass, if associated with irregular deposits of bone or calcification, is especially characteristic. Areas of lysis instead of the uniform calcification of osteochondroma is also suggestive of secondary chondrosarcoma. Computed tomograms and magnetic resonance images help in delineating the true thickness of the cartilage cap (Figs. 6.11 & 6.12).
GROSS PATHOLOGIC FEATURES
Chondrosarcomas may be divided into central and peripheral types. In long bones, this separation in type is usually obvious, with a rare peripheral sarcoma arising either on an osteochondroma or directly from the surface of a bone. The latter, a periosteal chondrosarcoma, is exceedingly rare, with only 17 examples in the Mayo Clinic files. Seven others were situated peripherally and did not have features of periosteal chondrosarcoma. They may have arisen in exostoses, but this could not be proven. The size of the lesion is most helpful in differentiating periosteal chondroma from chondrosarcoma. The former is almost always less than 3 cm in greatest dimension, whereas the latter is rarely less than 5 cm. If exostosis is present, the finding of a cartilaginous cap, irregularly thickened to more than 1 cm, must be viewed with the suspicion of malignancy; cartilaginous masses of 3 or 4 cm usually are indicative of a chondrosarcoma. In thin or flat bones, as in the pelvic girdle or thoracic cage, landmarks are so destroyed by the time the average tumor is diagnosed that the exact site of origin can only be surmised, but most of the tumors apparently begin centrally. As seen radiographically, a central chondrosarcoma often produces expansion and concomitant thickening of the cortex of long bones. In such cases, the region of involved marrow is usually distinctly demarcated. The thickened cortex is invaded by tumor, and breakthrough eventually occurs (Figs. 6.13 & 6.14).
Chondrosarcomas are characteristically composed of lobules that vary from a few millimeters to several centimeters in greatest dimension. Except at the periphery of the tumor, these lobules are usually completely coalesced. The centers of the lobules often become necrotic, liquefied, and cystic. The liquefaction and cystification are secondary to marked myxoid change of the matrix. The myxoid change may also give rise to a gelatinous appearance of the neoplasm. Necrotic foci often calcify in an irregular fashion. Some of the calcific zones observed grossly are actually osseous masses (Figs. 6.15, 6.16, 6.17, 6.18, 6.19 and 6.20).
Chondrosarcomas produce a matrix substance that varies in consistency from that of firm hyaline cartilage to that of mucus. A myxoid quality is an ominous sign, strongly suggestive of malignancy. Sometimes the periphery of the recurrent form of a cartilaginous tumor is opaque and fibrous, resembling a fibrosarcoma or even an osteosarcoma grossly and microscopically (Fig. 6.21).
It is extremely unusual to see a multicentric chondrosarcoma. There were only eight examples in the Mayo Clinic files. One patient had separate chondrosarcomas involving the rib and the sphenoid bone 2 years apart. One patient had a lesion of one distal femur and one distal fibula 5 years apart. Two patients had Ollier disease: one of them had a lesion of the ischium and 13 years later a chondrosarcoma of the proximal tibia, and the other had a chondrosarcoma of the humerus 7 years after an above-knee amputation for chondrosarcoma of the proximal tibia. One patient with multiple exostoses developed a chondrosarcoma of the sacrum 4 years after undergoing internal hemipelvectomy for a chondrosarcoma of the pubis. One patient had two chondrosarcomas involving the femur, and another patient had two chondrosarcomas involving the proximal and distal tibia.
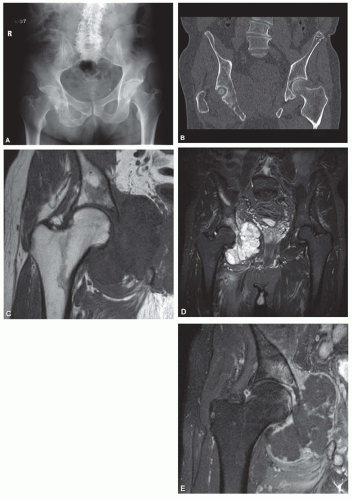 Figure 6.8. Anteroposterior radiograph (A) and two-dimensional coronal computed tomogram (B) of the pelvis in a 70-year-old man shows an aggressive destructive mixed lytic and sclerotic lesion in the right acetabulum and ischium, with scattered foci of chondroid matrix and an associated pathologic fracture. The coronal T1- (C) and T2- (D) weighted magnetic resonance images show that the lesion is associated with a large extraosseous component. The gadolinium-enhanced image (E) shows that there are large regions with little to no enhancement within the lesion, suggestive of myxoid change. The constellation of findings is most consistent with chondrosarcoma. Because of the large unmineralized soft-tissue mass, the differential diagnosis would also include dedifferentiated chondrosarcoma. |
 Figure 6.9. Chondrosarcoma arising in the setting of Ollier disease. A: Anteroposterior radiograph of the left hand shows multiple lytic lesions in the phalanges, with varying degrees of expansion typical of multiple enchondromas in a patient with Ollier disease. B: Sagittal T2-weighted magnetic resonance imaging with fat suppression shows cortical destruction and a large soft-tissue mass associated with one of the cartilage lesions in the fifth middle phalanx, consistent with chondrosarcoma arising in an enchondroma. The soft-tissue extension is not detectable in the radiograph. |
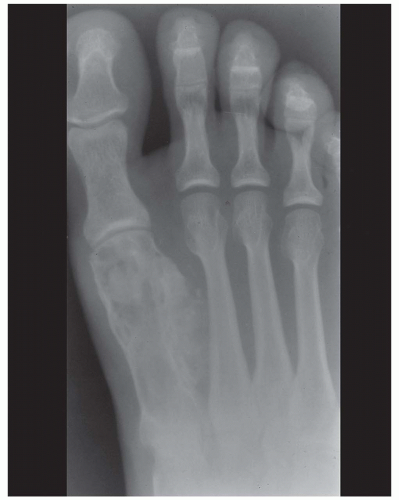 Figure 6.10. Chondrosarcoma of the first metatarsal in a 32-year-old man. The tumor extends into soft tissues. |
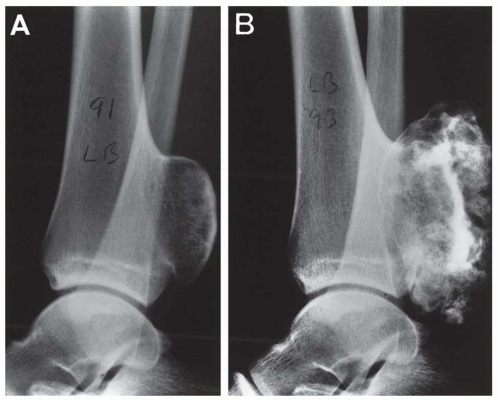 Figure 6.11. Osteochondroma of the distal tibia in an 18-year-old woman. A: The surface is smooth, and the cartilage cap is thin. B: Three years later, the cap is irregular and nodular, a change supporting the diagnosis of secondary chondrosarcoma. (Case provided by Dr. Cynthia Flessner, Memorial Medical Center, Springfield, Illinois.) |
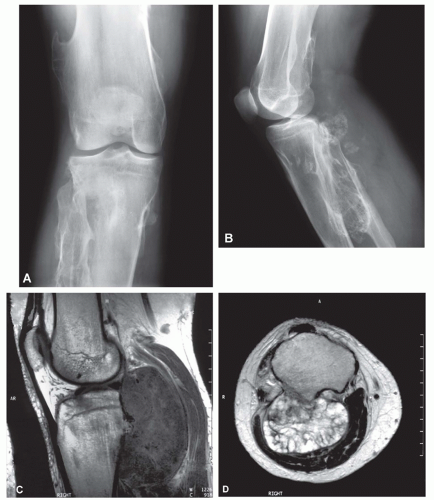 Figure 6.12. A: Anteroposterior radiograph shows multiple osteochondromas involving the bones of the knee, with associated developmental deformity compatible with multiple hereditary osteochondromatosis. B: Lateral radiograph shows malignant destruction of one of the lesions involving the proximal tibia posteriorly, with a large associated soft-tissue mass that contains flecks of cartilaginous matrix. Sagittal T1- (C) and axial T2- (D) weighted images show the large soft-tissue mass surrounding a remnant of the stalk of the osteochondroma. These imaging features are characteristic of malignant degeneration of an osteochondroma to chondrosarcoma. |
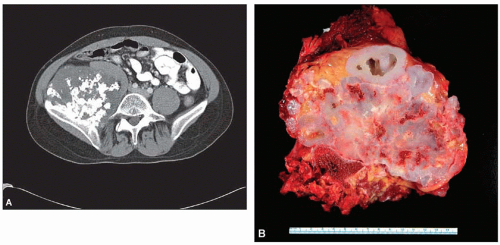 Figure 6.13. Secondary chondrosarcoma arising from a sessile osteochondroma in the right ilium of a 21-year-old man. A: Computed tomography shows a large mineralized mass. B: The lesion consists almost entirely of cartilage. There is gross cystic change. The cystic cavities contain gelatinous material representing myxoid change. |
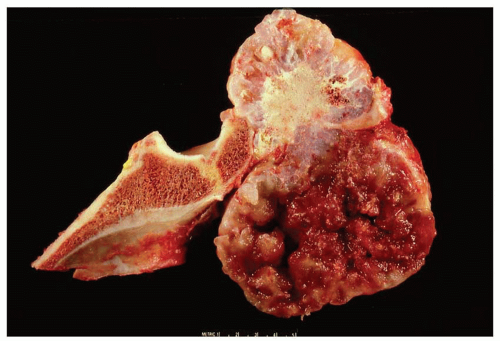 Figure 6.14. Secondary chondrosarcoma involving the ilium. The top portion of the field shows features of osteochondroma. The lower portion shows pure cartilaginous growth with marked myxoid change. |
 Figure 6.15. Grade 1 chondrosarcoma involving the right anterior lower seventh rib in a 48-year-old man. He presented with right upper quadrant pain, likely from this large chest wall mass that was abutting the right colon. |
Metastasis to regional lymph nodes is distinctly rare. In chondrosarcoma, hematogenous dissemination to the lungs is much less common than in osteosarcoma or fibrosarcoma.
Chondrosarcoma has a pronounced propensity for local recurrence, even when the surgeon has apparently removed the tumor completely.
HISTOPATHOLOGIC FEATURES
Chondrosarcoma may be difficult to diagnose purely on the basis of histologic features. The criteria that differentiate low-grade chondrosarcoma from a chondroma are very subtle. Experience has shown that different
rules apply in different clinical situations. Almost without exception, cartilaginous tumors of the sternum are malignant, regardless of the histologic appearance. However, some lesions may appear to be “malignant” by conventional criteria but are benign: chondromas of the small bones of the hands and feet, periosteal chondromas, chondromas of Ollier disease and Maffucci syndrome, synovial chondromatosis, and chondromas of the soft tissues of the hands and feet are examples. The features proposed by Lichtenstein and Jaffe are very helpful when viable fields are studied. These include many cells with plump nuclei, more than an occasional cell with two such nuclei, and, especially, giant cells with large single or multiple nuclei or clumps of chromatin. The correct diagnosis depends on the correct interpretation of the subtle qualitative characteristics. Furthermore, malignant foci may be obscured by necrotic regions or by zones with insufficient cytologic evidence for diagnosis of sarcoma, further complicating the pathologist’s task.
rules apply in different clinical situations. Almost without exception, cartilaginous tumors of the sternum are malignant, regardless of the histologic appearance. However, some lesions may appear to be “malignant” by conventional criteria but are benign: chondromas of the small bones of the hands and feet, periosteal chondromas, chondromas of Ollier disease and Maffucci syndrome, synovial chondromatosis, and chondromas of the soft tissues of the hands and feet are examples. The features proposed by Lichtenstein and Jaffe are very helpful when viable fields are studied. These include many cells with plump nuclei, more than an occasional cell with two such nuclei, and, especially, giant cells with large single or multiple nuclei or clumps of chromatin. The correct diagnosis depends on the correct interpretation of the subtle qualitative characteristics. Furthermore, malignant foci may be obscured by necrotic regions or by zones with insufficient cytologic evidence for diagnosis of sarcoma, further complicating the pathologist’s task.
 Figure 6.16. Grade 1 chondrosarcoma involving the pelvis. The tumor has gray-white lobulated areas corresponding to cartilaginous differentiation. Yellow areas of necrosis and degenerative myxoid change are also present. |
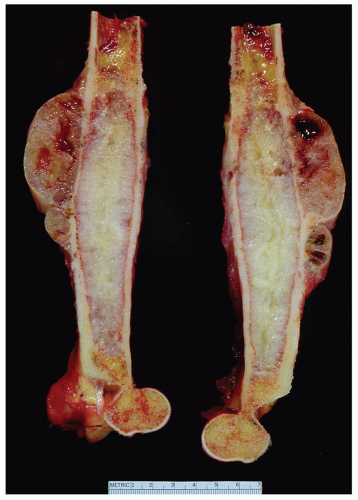 Figure 6.17. Chondrosarcoma involving the distal humerus. On radiographs, a large soft-tissue mass suggested a diagnosis of dedifferentiation. Microscopically, however, the entire tumor was a grade 2 chondrosarcoma. |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


