Chapter 74 Carnitine
 Introduction
Introduction
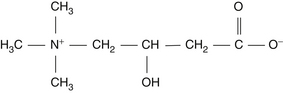
Carnitine is an essential nutrient for the transport of long-chain fatty acids into the mitochondrial matrix. Carnitine (β-hydroxy/γ-butyrobetaine) was originally isolated from meat extracts in 1905, and its exact chemical structure was determined in 1932 (Figure 74-1). However, despite extensive physiologic and pharmacologic studies in the 1930s, no physiologic role for carnitine was determined until 1955.1–3
The compound was virtually forgotten until Carter et al4 created new interest in carnitine in 1952, when they established it as a growth factor for the meal worm Tenebrio molitor (hence carnitine’s other name, “vitamin BT”—B for the family of vitamins and T for Tenebrio molitor). When other species of organisms were also shown to be dependent on carnitine, researchers began to reexamine its role in humans.
Researchers soon found that carnitine was essential in the oxidation of lipids.1–3 When the first carnitine-deficient human subjects were described in 1973, it stimulated greater investigation.5
 Biosynthesis
Biosynthesis
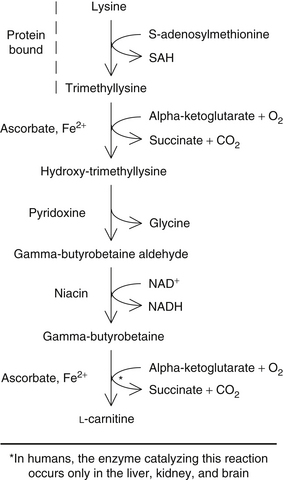
Carnitine is synthesized in humans from lysine and methionine. In nonmammalians, carnitine synthesis begins with stepwise methylation of free lysine by S-adenosylmethionine to produce trimethyllysine. In mammals, however, protein-bound trimethyllysine, rather than free lysine, appears to be the major precursor for carnitine synthesis.1–3
Trimethyllysine is then converted through a series of enzymatic reactions (occurring in the liver, kidney, brain, heart, and skeletal muscle) to butyrobetaine. However, the conversion of butyrobetaine to carnitine can only occur in the liver, kidney, and brain, since the enzyme required, butyrobetaine hydroxylase, is only present in these tissues.1–3
The synthesis of carnitine is largely controlled by the activity of butyrobetaine hydroxylase. This enzyme appears to be age dependent. In infancy, the activity of butyrobetaine hydroxylase has been shown to be only 12% of the normal adult mean. By 2.5 years, the activity is 30% of the adult mean, and by 15 years, the level is within a standard deviation of the adult mean.1–3 These data would seem to indicate the importance of preformed carnitine in breast milk.
As apparent from Figure 74-2, two essential amino acids (lysine and methionine), three vitamins (ascorbate, niacin, and vitamin B6), and a metal ion (reduced iron) are required for the synthesis of carnitine. Obviously, a deficiency of any one of these nutrients would result in significantly impaired carnitine synthesis.1–3 However, research using knockout mice unable to synthesize vitamin C endogenously showed normal carnitine values (comparable to their vitamin C synthesizing counterparts), although they were vitamin C deficient. The researchers suggested that adequate levels of glutathione and glutathione peroxidase might compensate or replace the deficient vitamin C in the carnitine biosynthesis pathway.6
 Metabolism
Metabolism
Pharmacokinetics
The heart and skeletal muscles, as well as many other tissues, depend primarily on fatty acid oxidation as a source of energy. Because they cannot synthesize carnitine, its transport into these tissues is of critical importance.
Specific carnitine-binding transport proteins were identified for several tissues (e.g., cardiac muscle, skeletal muscle, epididymis, liver, kidney) taking the serum carnitine into the cell.1,3 Through this active transport mechanism, the tissues can concentrate carnitine up to 10 times greater than that found in plasma.
A small in vivo study measured the pharmacokinetics of carnitine in Chinese men and women participants. A 2000 mg single oral dose of L-carnitine (LC) had a half-life of approximately 60.3 hours compared with L-acetylcarnitine (LAC) at 35.9 hours and L-propionylcarnitine (LPC) at 25.7 hours. The maximum plasma concentration of LC, LAC, and LPC occurred at 3.4, 2.4, and 3.8 hours after ingestion, respectively, and at quantities of 84.7, 12.9, and 5.1 µmol/L, respectively. The researchers found comparable levels of carnitine absorption and metabolism between men and women, suggesting that gender should not affect dosage.7
Urinary excretion of unchanged carnitine is the major route of elimination of carnitine. Since the tubular reabsorption of carnitine by the kidneys is extremely efficient, the daily turnover of carnitine is estimated to be only 4% to 6% of the total body pool of the healthy individual.1–37 Factors that increase carnitine excretion and degradation are discussed later in the section on “Deficiency.”
 Physiologic Functions
Physiologic Functions
Carnitine’s basic function is in the transport of long-chain fatty acids into the mitochondrial matrix and the facilitation of β-oxidation.13 Because acyl coenzyme A formed in the endoplasmic reticulum or outer mitochondrial membrane cannot penetrate the inner mitochondrial membrane to the site of fatty acid β-oxidation, the acyl group must be transferred from coenzyme A to carnitine. The acyl-carnitine molecule then transports the fatty acid molecule to the mitochondrial surface of the inner mitochondrial membrane and releases the fatty acid into the matrix, where β-oxidation occurs. Figure 74-3 summarizes this process.
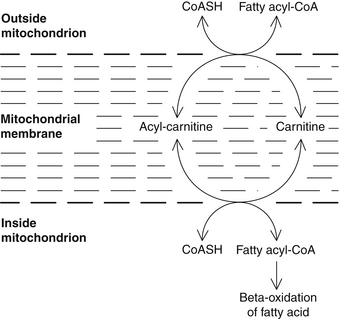
FIGURE 74-3 Role of carnitine in the transport of long-chain fatty acids through the inner mitochondrial membrane.
Carnitine has several other physiologic functions, including oxidation of the ketoacid analogs of the branched chain amino acids valine, leucine, and isoleucine.1–3 This function is extremely important during fasting, starvation, and exercise.
 Deficiency
Deficiency
Carnitine deficiency may arise from several causes, as listed in Box 74-1.
BOX 74-1 Causes of Carnitine Deficiency
• Dietary deficiency of the precursor amino acids lysine and methionine
• Deficiency of any cofactor (e.g., iron, ascorbic acid, pyridoxine, niacin) required by the enzymes of the lysine to carnitine pathway
• Genetic defect of carnitine biosynthesis
• Defective intestinal absorption of carnitine
• Liver or kidney dysfunction that impairs carnitine synthesis
• Increased metabolic losses of carnitine due to catabolism, impaired tubular resorption, or genetic defect
• Defective transport of carnitine from tissues of synthesis to tissues where it is maximally used
• Increased carnitine requirement due to a high-fat diet, drugs (e.g., valproic acid), metabolic stress, or disease
Carnitine deficiency states have been classified into two major groups:
Diagnosis of systemic carnitine deficiency can be made using serum or 24-hour urine samples. Total, free, and esterified carnitine levels should be determined. In myopathic carnitine deficiency, diagnosis requires skeletal muscle biopsy.8
To date, no patients with primary systemic carnitine deficiency have been identified. The systemic deficiency has always been secondary to some other factor rather than a defect in carnitine synthesis.1–3,8,9
The consequences of systemic carnitine deficiency are impaired lipid metabolism and lipid accumulation in the skeletal muscles, myocardium, and liver. Progressive muscle weakness with lipid storage myopathy is found in all patients.1–38 In adults, auxiliary nonmitochondrial oxidation mechanisms are apparently stimulated, resulting in some degree of adaptation. This adaptation occurs in starvation, diabetes, high-fat diets, and other causes of secondary carnitine deficiency. Systemic carnitine deficiencies usually respond dramatically to orally administered supplemental LC.8,9
Children are apparently unable to adapt to low carnitine levels as well as adults.8 Several cases of carnitine deficiency in children, presenting a clinical picture resembling Reye’s syndrome (acute encephalopathy associated with altered liver function due to lipid accumulation), have been reported.9–11
The clinical presentation of secondary carnitine deficiency in children includes hypotonia, failure to thrive, recurrent infections, encephalopathy, nonketotic hypoglycemia, and cardiomyopathy.8 Several fatal cases of systemic carnitine deficiency have been reported.9,12
In primary myopathic carnitine deficiency, there is an inborn error of carnitine metabolism that is limited to skeletal muscle.8,9 The defect appears to be in the transport of carnitine into the skeletal muscle as serum carnitine; the carnitine levels in other tissues are normal. Severe lipid storage myopathy is the result. Supplemental carnitine is generally of no value in myopathic carnitine deficiency. Rather, improvements have been noted using diets high in medium-chain triglycerides and low in long-chain triglycerides.8
 Carnitine as a Nutrient
Carnitine as a Nutrient
Analysis of several hundred foods for carnitine content indicates that meat and dairy products are the major dietary sources of carnitine.3 In general, the redder the meat, the higher the carnitine content. Cereals, fruits, and vegetables contain little or no carnitine. Preliminary studies indicate that the daily diet contains 5 to 100 mg of carnitine.3
Carnitine in the Infant Diet
Oxidation of long-chain fatty acids, which requires carnitine, is well known to be critical to the survival and normal development of the newborn.3 Carnitine concentrations in fetal and umbilical cord blood are higher than in maternal blood, suggesting the placenta may actively transport carnitine to the fetus, because carnitine synthesis is not fully developed.3 The initial carnitine concentration in the newborn depends on maternal carnitine concentration.
Supplementation of carnitine during pregnancy may be necessary to ensure adequate tissue concentrations in the fetus, as well as the mother. Serum carnitine levels are typically lower in pregnant women than nonpregnant women, presumably due to increased excretion.13,14
The newborn infant is almost entirely dependent on external sources of carnitine.3 Breast-fed infants have the best chance of achieving optimal carnitine concentrations. The bioavailability of carnitine from breast milk is significantly greater than that in cow’s milk–based formulas,15 and soy-based infant formulas contain no detectable carnitine.3 Formula feeding may necessitate supplemental carnitine to achieve normal carnitine concentrations in these infants.
Carnitine administration to preterm infants has potentiated weight gain and growth.16 In preterm infants, serum values of carnitine decrease dramatically due to limited storage capacity coupled with a decreased ability to synthesize carnitine. Administration of LC to preterm infants is considered important.
 Clinical Applications
Clinical Applications
Many disease states, in addition to classic as well as secondary carnitine deficiency, may benefit from carnitine administration. Evidence supports the assertion that supplemental carnitine may benefit the conditions listed in Box 74-2 and discussed later.
BOX 74-2 Conditions That May Benefit from Carnitine Supplementation
• Arrhythmias and cardiotoxicity induced by drugs
• Familial endocardial fibroelastosis
• Idiopathic mitral valve prolapse
• Elevated triglyceride levels
• Enhancing physical performance
• Fatigue in person with celiac disease
• Alzheimer’s disease, senile depression, and age-related memory defects
• Kidney disease and hemodialysis
• Alcohol-induced fatty liver disease
• Low sperm counts and decreased sperm motility
• Chronic obstructive pulmonary disease
• Acquired immunodeficiency syndrome
• Inborn errors of amino acid metabolism
Carnitine is available in several different forms. The form being used must be LC alone or bound to either acetic (LAC) or propionic acid (LPC). The D form of carnitine (discussed later in the section on “Toxicology”) should never be used. The reason for using carnitine will dictate the form used. For Alzheimer’s disease and brain effects, it appears that LAC may provide the greatest benefit. For angina, LPC may be the best choice, because the myocardium appears to prefer it to LAC, followed by LC.17,18 LC is, however, the most widely available, least expensive, and best studied form of carnitine.
Cardiovascular Disease
Oral LAC therapy (1 g twice daily) for 6 months showed a gradual but significant decrease in blood pressure in insulin resistant participants who had higher risk factors for cardiovascular disease (hypertension, hypertriglyceridemia, obesity, and/or family history of diabetes type 2). In the patients with greater insulin resistance, the insulin sensitivity was significantly increased. The overall adiponectin values increased. These benefits were slowly reversed during an 8-week follow-up after LAC discontinuation. There were no changes to participants’ diet or lifestyle during this study. LAC was well tolerated in all participants.19 It appears that LAC can improve aspects that are characteristic of metabolic syndrome, but without lifestyle and dietary modifications, the patient may potentially need long-term supplementation with carnitine.
Carnitine exerts a beneficial effect on blood lipids by lowering triglycerides (TGs) and total cholesterol levels while raising high-density lipoprotein (HDL) cholesterol. After 4 months of therapy with LC in patients with elevated blood lipids, typical changes observed were a 20% reduction for total cholesterol, a 28% decrease in TGs, and a 12% increase in HDL levels.20,21 Due to the higher cost of carnitine compared with other natural agents (e.g., inositol hexaniacinate, garlic, and gugulipid), its use should be reserved for those cases unresponsive to these more cost-effective measures.
Angina and Recovery from Myocardial Infarction
Carnitine is useful in angina due to its ability to improve oxygen usage and energy metabolism by the myocardium. As a result of improving fatty acid usage and energy production, carnitine also prevents the production of toxic fatty acid metabolites.22 These compounds are extremely damaging, because they disrupt cellular membranes. Changes in the properties of cell membranes throughout the heart are thought to contribute to impaired contraction of the heart muscle, increased susceptibility to irregular beats, and eventual death of heart tissue. Supplementing with carnitine increases heart carnitine levels and has been shown to prevent the production of toxic fatty acid metabolites, as well as boost antioxidant enzyme levels. In addition to angina, all of these effects make carnitine beneficial in recovery from a heart attack, cardiomyopathies, arrhythmias, and congestive heart failure.23,24
Numerous clinical trials demonstrated that carnitine improves angina and heart disease (note that all three commercial forms have been used).23,25–32 Improvements were noted in exercise tolerance and heart function. The results indicated that carnitine is an effective alternative to drugs in cases of angina.
LPC may offer the greatest benefit in angina, as well as in other cardiovascular conditions. LPC is taken up by myocardial cells much more rapidly than other forms of carnitine.17 In one study, LPC (15 mg/kg intravenously) significantly diminished myocardial ischemia as demonstrated by a significant 12% and 50% reduction in ST-segment depression and left ventricular end-diastolic pressure, respectively, during the atrial pacing test.33 Left ventricular ejection fraction increased by 18%. Recovery of heart function after exercise occurred much quicker in the LPC group compared with the placebo group.
LC and LAC also showed good results. In one of the larger studies, 200 patients with exercise-induced stable angina received either standard therapy alone (e.g., nitroglycerin, calcium channel blockers, β-blockers, antihypertensives, diuretics, digitalis, antiarrhythmics, anticoagulants, hypolipidemics) or in combination with 2000 mg/day of LC over a 6-month period.34 Compared with the control group, the patients on LC exhibited a significant reduction in premature ventricular contractions at rest, as well as an increased tolerance to exercise as demonstrated by an increased maximal cardiac frequency, increased maximal systolic blood pressure, cardiac output, and reduced ST-segment depression (70% reduction in the LC group versus no change in the control group). Reductions in low-density lipoprotein (LDL) cholesterol (8%) and TGs (12%) were also noted. These results are highly significant and provide a strong rationale for the inclusion of carnitine in patients using standard medical therapy.
In Italy, a large study involving 472 patients showed additional benefits.35 The study was performed to evaluate the effects of LC administration on chronic left ventricular dilation in patients with acute anterior myocardial infarction. Placebo or LC was given at a dose of 9 g/day intravenously for the first 5 days and then 6 g/day orally for the next 12 months. Left ventricular volumes and ejection fraction were evaluated on admission, at discharge from hospital, and at 3, 6, and 12 months after acute myocardial infarction. A significant attenuation of left ventricular dilation in the first year after acute myocardial infarction was observed in patients treated with LC compared with those receiving placebo. The percent increase in both end-diastolic and end-systolic volumes from admission to 3-, 6-, and 12-month evaluation was significantly reduced in the LC group.
Congestive Heart Failure
Several double-blind clinical studies showed that carnitine (again, LPC appeared to be more effective than LC or LAC) improved cardiac function in patients with congestive heart failure.23,36,37 In one double-blind study of LPC versus placebo in a group of 60 patients with mild to moderate (New York Heart Association [NYHA] classes II and III) congestive heart failure, LPC produced demonstrable benefit.36 The group was made up of men and women between 48 and 73 years old receiving long-term treatment with digitalis and diuretics for at least 3 months and who still displayed symptoms. Thirty of these patients were chosen randomly and for 180 days received 500 mg of LPC three times a day in addition to their usual treatment. At basal conditions and after 30, 90, and 180 days, the maximum exercise time was evaluated using an exercise tolerance test performed on an ergometer bicycle, and the left ventricular ejection fraction was tested by echocardiography. After 1 month of treatment, the patients treated with LPC, compared with the control group, showed significant increases in the values of both tests, increases that became even more evident after 90 and 180 days. At the stated times, the increases in the maximum exercise time were 16.4%, 22.9%, and 25.9%, respectively. The ventricular ejection fraction increased by 8.4%, 11.6%, and 13.6%, respectively.
Even more obvious benefits were seen in a 3-year study of 80 patients with moderate to severe heart failure (NYHA class III to IV) caused by dilated cardiomyopathy. After a period of stable cardiac function up to 3 months, patients were randomly assigned to receive either carnitine (2 g/day orally) or placebo. After a mean of 33.7 months of follow-up (range 10 to 54 months), 70 patients remained in the study: 33 in the placebo group and 37 in the carnitine group. At the time of analysis, 63 patients were alive. Six deaths occurred in the placebo group, and one death in the carnitine group. Survival analysis showed that patients’ survival was statistically significant in favor of the carnitine group.38

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


