Chapter 2 Cardiac Ion Channels
Ion channels are pore-forming membrane proteins that regulate the flow of ions passively down their electrochemical gradient across the membrane. Ion channels are present on all membranes of cells (plasma membrane) and intracellular organelles (nucleus, mitochondria, endoplasmic reticulum). There are more than 300 types of ion channels in a living cell. The channels are not randomly distributed in the membrane, but tend to cluster at the intercalated disc in association with modulatory subunits.1
Importantly, channel opening and closing are not instantaneous but usually take time. The transition from the resting (closed) state to the open state is called activation. Once opened, channels do not remain in the open state, but instead they undergo conformational transition in a time-dependent manner to a stable nonconducting (inactivated) state. Inactivated channels are incapable of reopening and must undergo a recovery or reactivation process back to the resting state to regain their ability to open. Inactivation curves of the various voltage-gated ion channel types differ in their slopes and midpoints of inactivation and can overlap, in which case a steady-state or noninactivating current flows.1
Ion channels differ with respect to the number of subunits of which they are composed and other aspects of structure. Many ion channels function as part of macromolecular complexes in which many components are assembled at specific sites within the membrane. For most ion channels, the pore-forming subunit is called the α subunit, whereas the auxiliary subunits are denoted β, gamma, and so on. Most ion channels have a single pore; however, some have two.1
Sodium Channels
Structure and Physiology
The cardiac Na+ channel complex is composed of a primary α and multiple ancillary β subunits. The approximately 2000-amino-acid α subunit contains the channel’s ion-conducting pore and controls the channel selectivity for Na+ ions and voltage-dependent gating machinery. This subunit contains all the drug and toxin interaction sites identified to date. The α subunit (Nav1.5), encoded by the SCN5A gene, consists of four internally homologous domains (I to IV) that are connected to each other by cytoplasmic linkers (Fig. 2-1). Each domain consists of six membrane-spanning segments (S1 to S6), connected to each other by alternating intracellular and extracellular peptide loops. The four domains are arranged in a fourfold circular symmetry to form the channel. The extracellular loops between S5 and S6 (termed the P segments) have a unique primary structure in each domain (Fig. 2-2). The P segments curve back into the membrane to form an ion-conducting central pore whose structural constituents determine the selectivity and conductance properties of the Na+ channel.2
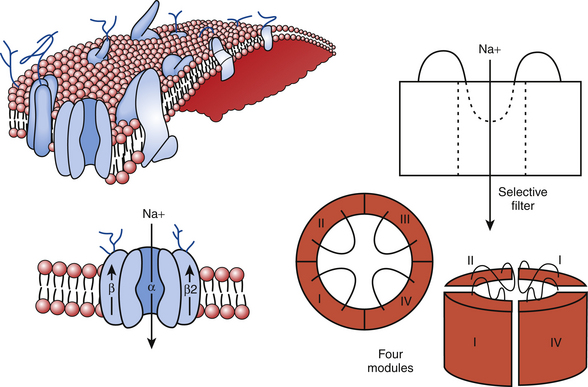
FIGURE 2-1 The sodium channel macromolecular complex. See text for discussion.
(From Boussy T, Paparella G, de Asmundis C, et al: Genetic basis of ventricular arrhythmias. Heart Fail Clin 6:249-266, 2010.)
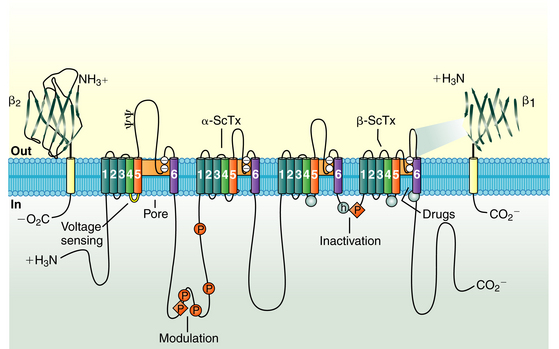
Four auxiliary β subunits (Navβ1 to Navβ4, encoded by the genes SCN1B to SCN4B, respectively) have been identified; each is a glycoprotein with a single membrane-spanning segment. The β1 subunit likely plays a role in modulation of the gating properties and level of expression of the Na+ channel.2
Na+ channels are the typical example of voltage-gated ion channels. Na+ channels switch among three functional states: deactivated (closed), activated (open), and inactivated (closed), depending on the membrane potential (Em). These channel states control Na+ ion permeability through the channel into the cardiomyocyte. Na+ channel activation allows Na+ ion influx into the cell, and inactivation blocks entry of Na+ ions.2
On excitation of the cardiomyocyte by electrical stimuli from adjacent cells, its resting Em (approximately −85 mV) depolarizes. The positively charged S4 segment of each domain of the α subunit functions as the sensor of the transmembrane voltage; these segments are believed to undergo rapid structural conformational changes in response to membrane depolarization, thus leading to channel opening (activation) from its resting (closed) state and enabling a large and rapid influx of Na+ (inward Na+ current [INa]) during the rapid upstroke (phase 0) of the action potential in atrial, ventricular, and Purkinje cardiomyocytes.3
Normally, activation of Na+ channels is transient; fast inactivation (closing of the pore) starts simultaneously with activation, but because inactivation is slightly delayed relative to activation, the channels remain transiently open to conduct INa during phase 0 of the action potential before it closes. Each Na+ channel opens very briefly (less than 1 millisecond) during phase 0 of the action potential; collectively, activation of the channel lasts a few milliseconds and is followed by fast inactivation.
Na+ channel inactivation comprises different conformational states, including fast, intermediate, and slow inactivation. Fast inactivation is at least partly mediated by rapid occlusion of the inner mouth of the pore by the cytoplasmic interdomain linker between domains III and IV of the α subunit, which has a triplet of hydrophobic residues that likely functions as a hinged “latch” that limits or restricts Na+ ion pass through the pore. The carboxyl terminus (C-terminus) also plays an important role in the control of Na+ channel inactivation and stabilizing the channels in the inactivated state by interacting with the loop linking domains III and IV. Importantly, although most Na+ channels open before inactivating, some actually inactivate without ever opening (a process known as closed-state inactivation).1,4
In addition to these rapid gating transitions, Na+ channels are also susceptible to slower inactivating processes (slow inactivation) if the membrane remains depolarized for a longer time. These slower events can contribute to the availability of active channels under various physiological conditions. Whereas fast-inactivated Na+ channels recover rapidly (within 10 milliseconds) during the hyperpolarized interval between stimuli, slow inactivation requires much longer recovery times (ranging from hundreds of milliseconds to many seconds). The molecular movements leading to slow inactivation are less well understood. The P segments seem to play a key role in slow inactivation.4
Some Na+ channels occasionally show alternative gating modes consisting of isolated brief openings occurring after variable and prolonged latencies and bursts of openings during which the channel opens repetitively for hundreds of milliseconds. The isolated brief openings are the result of the occasional return from the inactivated state. The bursts of openings are the result of occasional failure of inactivation.1 Prolonged opening or reopening of some Na+ channels during phases 2 and 3 can result in a small late INa (INaL). Despite its minor contribution in healthy hearts, INaL can potentially play an important role in diseased hearts.3
Function
The cardiac Na+ channel also plays a crucial role in the propagation of action potentials throughout the atrium, HPS, and ventricles. The opening of Na+ channels in the atria underlies the P wave on the ECG, and in the ventricles INa underlies the QRS complex and enables a synchronous ventricular contraction. Because the upstroke of the electrical potential primarily determines the speed of conduction between adjacent cells, Na+ channels are present in abundance in tissues where speed is of importance. Cardiac Purkinje cells contain up to 1 million Na+ channels, a finding that illustrates the importance of rapid conductance in the heart.2
Na+ channels also make a contribution in the plateau phase (phase 2) and help determine the duration of the action potential. After phase 0 of the action potential, INa decreases to less than 1% of its peak value over the next several milliseconds because of voltage-dependent inactivation. This persistent or “late” inward INa (INaL), along with the L-type Ca2+ current (ICaL), helps maintain the action potential plateau.5
Regulation
The regulatory proteins interacting with Nav1.5 may be classified as follows: (1) anchoring-adaptor proteins (e.g., ankyrin-G, syntrophin proteins, multicopy suppressor of gsp1 [MOG1]), which play roles only in trafficking and targeting the channel protein in specific membrane compartments; (2) enzymes interacting with and modifying the channel structure (post-translational modifications), such as protein kinases or ubiquitin ligases; and (3) proteins modulating the biophysical properties of Nav1.5 on binding (e.g., caveolin-3, calmodulin, glycerol 3-phosphate dehydrogenase 1–like [G3PD1L], telethonin, Plakophilin-2).5 Coexpression of Nav1.5 with its β subunits induces acceleration in the recovery from inactivation and enhancement of INa amplitude.
The cardiac Na+ channels are subject to phosphorylation and dephosphorylation by kinases or phosphatases. The intracellular linker between domains I and II contains eight consensus sites for cyclic adenosine monophosphate (cAMP)–dependent protein kinase A (PKA) phosphorylation. cAMP-dependent PKA and G protein stimulatory α subunit (Gsα) modulate the function of expressed cardiac Na+ channels on β-adrenergic stimulation and enhance INa.1
In contrast, activation of α-adrenergic stimulating protein kinase C (PKC) results in the reduction of INa. The effect of PKC is largely attributable to phosphorylation of a highly conserved serine in the linker between domains III and IV. PKC reduces the maximal conductance of the channels and alters gating. Na+ channels exhibit a hyperpolarizing shift in the steady-state availability curve, suggesting an enhancement of inactivation from closed states.
All subunits of the Na+ channel are modified by glycosylation. The β1 and β2 subunits are heavily glycosylated, with up to 40% of the mass being carbohydrate. In contrast, the cardiac α subunit is only 5% sugar by weight. Sialic acid is a prominent component of the N-linked carbohydrate of the Na+ channel. The addition of such a highly charged carbohydrate has predictable effects on the voltage dependence of gating through alteration of the surface charge of the channel protein.1
Pharmacology
Na+ channels are the targets for the action of class I antiarrhythmic drugs. Na+ channel blockers bind to a specific receptor within the channel’s pore. The binding blocks ion movement through the pore and stabilizes the inactivated state of Na+ channels. Blockade of Na+ channels tends to decrease tissue excitability and conduction velocity (by attenuating peak INa) and can shorten action potential duration (by attenuating late INa).1,6
One important component in the action of antiarrhythmic drugs is a voltage-dependent change in the affinity of the drug-binding site (i.e., the channel is a modulated receptor). Additionally, restricted access to binding sites can contribute to drug action, a phenomenon that has been called the guarded receptor model. Open and inactivated channels are more susceptible to block than resting channels, likely because of a difference in binding affinity or state-dependent access to the binding site. Consequently, binding of antiarrhythmic drug occurs primarily during the action potential (known as use-dependent block), and the block dissipates after repolarization (i.e., in the interval between action potentials). When the time interval between depolarizations is insufficient for block to recover before the next depolarization occurs (secondary to either abbreviation of the interval between action potentials during fast heart rates or slow kinetics of the unbinding of the Na+ channel blocker), block of Na+ channels accumulates (resulting in an increased number of blocked channels and enhanced blockade).6 A drug with rapid kinetics produces less channel block with the subsequent depolarization than does a drug with slower recovery. Use-dependent block is important for the action of antiarrhythmic drugs because it allows strong drug effects during fast heart rates associated with tachyarrhythmias but limits Na+ channel block during normal heart rates. Importantly, drug recovery kinetics can potentially be slowed by pathophysiological conditions such as membrane depolarization, ischemia, and acidosis.1 This property is known as use-dependence and is seen most frequently with the class IC agents, less frequently with the class IA drugs, and rarely with the class IB agents.
Inherited Channelopathies
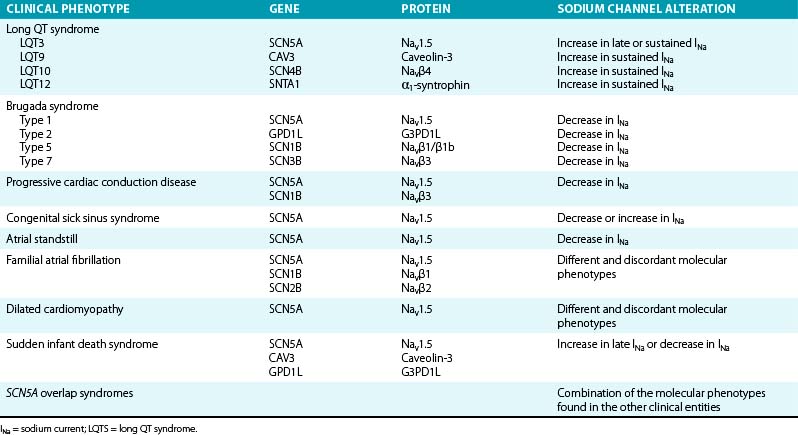
Mutations in genes that encode various subunits of the cardiac Na+ channel or proteins involved in regulation of the inward INa have been linked to several types of electrical disorders (Table 2-1). Depending on the mutation, the consequence is either a gain of channel function (with consequent prolongation of action potential duration because more positive ions accumulate in the cell) or an overall loss of channel function that influences the initial depolarizing phase of the action potential (with consequent decrease in cardiac excitability and electrical conduction velocity). It is noteworthy that a single mutation can cause different phenotypes or combinations thereof.2
Long QT Syndrome
In contrast to most long QT syndrome (LQTS) phenotypes, which are based on mutations that modify the cardiac K+ currents, type 3 congenital LQTS (LQT3), which accounts for approximately 8% of congenital LQTS cases, is caused by gain-of-function mutations on the Na+ channel gene, SCN5A. More than 80 mutations have been identified in the SCN5A gene, with most being missense mutations mainly clustered in Nav1.5 regions that are involved in fast inactivation (i.e., S4 segment of domain IV, the domain III–domain IV linker, and the cytoplasmic loops between the S4 and S5 segments of domain III and domain IV), or in regions that stabilize fast inactivation (e.g., the C-terminus).2,4,7
Several mechanisms have been identified to underlie ionic effects of SCN5A mutations in LQT3. Most SCN5A mutations cause a gain of function through disruption of fast inactivation, thus allowing repeated reopening during sustained depolarization and resulting in an abnormal, small, but functionally important sustained (or persistent) noninactivating Na+ current (Isus) during action potential plateau. Because the general membrane conductance is small during the action potential plateau, the presence of a persistent inward INa, even of small amplitude, can potentially have a major impact on the plateau duration and can be sufficient to prolong repolarization and QT interval. QT prolongation and the risk of developing arrhythmia are more pronounced at slow heart rates, when the action potential duration is longer, thereby allowing more INa to enter the cell.2,4,7
Other less common mechanisms of SCN5A mutations to cause LQT3 include increased window current, which results from delayed inactivation of mutant Na+ channels, occurring at more positive potentials and widening the voltage range during which the Na+ channel may reactivate without inactivation. Additionally, some mutations cause slower inactivation, which allows longer channel openings and causes a slowly inactivating INa. This current is INaL and is to be distinguished from Isus (which does not inactivate). Comparable to Isus, both the window current and INaL exert their effects during phases 2 and 3 of the action potential, in which normally no or very small INa is present. Other mutations induce prolonged action potential duration by enhancing recovery from inactivation, an effect that leads to larger peak INa by increasing the fraction of channels available for activation (because of faster recovery) during subsequent depolarizations. Finally, some mutations can cause increased expression of mutant Nav1.5 through enhanced mRNA translation or protein trafficking to the sarcolemma, decreased protein degradation, or altered modulation by β subunits and regulatory proteins. These effects lead to larger INa density during phase 0 of the action potential. Importantly, one single SCN5A mutation can potentially cause several changes in the expression and/or gating properties of the resulting Na+ channels.2
Regardless of the mechanism, increased Na+ current (Isus, window current, INaL, or peak INa) upsets the balance between depolarizing and repolarizing currents in favor of depolarization. The resulting delay in the repolarization process triggers early afterdepolarizations (EADs; i.e., reactivation of the L-type Ca2+ channel during phase 2 or 3 of the action potential), especially in Purkinje fiber myocytes, in which action potential durations are intrinsically longer.2
LQT9 is caused by gain-of-function mutations on the CAV3 gene, which encodes caveolin-3, a plasma membrane scaffolding protein that interacts with Nav1.5 and plays a role in compartmentalization and regulation of channel function. Mutations in caveolin-3 induce kinetic alterations of the Nav1.5 current that result in persistent Na+ current (Isus) and have been reported in cases of sudden infant death syndrome (SIDS).2,8
LQT10 is caused by loss-of-function mutations on the SCN4B gene, which encodes the β subunit (Navβ4) of the Nav1.5 channel. To date, only a single mutation in one patient has been described. This mutation caused a shift in the inactivation of the INa toward more positive potentials, but it did not change the activation. This resulted in increased window currents at an Em corresponding to the phase 3 of the action potential.2,8
LQT12 is caused by mutations on the SNTA1 gene, which encodes α1 syntrophin, a cytoplasmic adaptor protein that enables the interaction among Nav1.5, nitric oxide synthase, and the sarcolemmal Ca2+ adenosine triphosphatase (ATPase) complex that appears to regulate ion channel function. By disrupting the interaction between Nav1.5 and the sarcolemmal Ca2+ ATPase complex, SNTA1 mutations cause increased Nav1.5 nitrosylation with consequent reduction of channel inactivation and enhanced Isus densities.2,9
Brugada Syndrome
The Brugada syndrome is an autosomal dominant inherited channelopathy characterized by an elevated ST segment or J wave appearing in the right precordial leads. This syndrome is associated with a high incidence of sudden cardiac death (SCD) secondary to a rapid polymorphic ventricular tachycardia (VT) or ventricular fibrillation (VF). Approximately 65% of mutations identified in the SCN5A gene are associated with the Brugada syndrome phenotype (Brugada syndrome type 1), and they account for approximately 18% to 30% of cases of Brugada syndrome. So far, more than 200 Brugada syndrome–associated loss-of-function (i.e., reduced peak INa) mutations have been described in SCN5A. Some of these mutations result in loss of function secondary to impaired channel trafficking to the cell membrane (i.e., reduced expression of functional Na+ channels), disrupted ion conductance (i.e., expression of nonfunctional Na+ channels), or altered gating function. Altered gating properties comprise delayed activation (i.e., activation at more positive potentials), earlier inactivation (i.e., inactivation at more negative potentials), faster inactivation, and enhanced slow inactivation.1–3,10
Most of the mutations are missense mutations, whereby a single amino acid is replaced by a different amino acid. Missense mutations commonly alter the gating properties of mutant channels. Because virtually all reported SCN5A mutation carriers are heterozygous, mutant channels with altered gating may cause up to 50% reduction of INa. Different SCN5A mutations can cause different degrees of INa reduction and therefore different degrees of severity of the clinical phenotype of Brugada syndrome.10–12
In addition to SCN5A alterations, mutations in the GPD1L gene, which encodes the glycerol 3-phosphate dehydrogenase 1–like protein (G3PD1L), affect the trafficking of the cardiac Na+ channel to the cell surface and result in reduction of INa and Brugada syndrome type 2.10 Brugada syndrome associated with GPD1L gene mutations is characterized by progressive conduction disease, a low sensitivity to procainamide, and a relatively good prognosis.2,13
Furthermore, reduction in INa can be caused by mutations in the SCN1B gene (encoding the β1 and β1b subunits of the Na+ channel) and the SCN3B gene (encoding the β3 subunit of the Na+ channel), resulting in Brugada syndrome type 5 and type 7, respectively.10,13
Familial Progressive Cardiac Conduction Disease
Loss-of-function SCN5A mutations have been linked to familial forms of progressive cardiac conduction disease (referred to as hereditary Lenègre disease, primary cardiac conduction system disease, and familial atrioventricular [AV] block). This disease is characterized by slowing of electrical conduction through the atria, AVN, His bundle, Purkinje fibers, and ventricles, accompanied by an age-related degenerative process and fibrosis of the cardiac conduction system, in the absence of structural or systemic disease. It is often reflected by varying degrees of AV block and bundle branch block. Whether the age-dependent fibrosis of the conduction system is a primary degenerative process in progressive cardiac conduction disease or a physiological process that is accelerated by INa reduction remains to be investigated. A single loss-of-function SCN5A mutation can cause isolated progressive cardiac conduction disease or can be combined with the Brugada syndrome (overlap syndrome). Loss-of-function mutations in SCN1B also have been identified in patients with progressive cardiac conduction disease who carried no mutation in SCN5A.2
Congenital Sick Sinus Syndrome
Although INa does not play a prominent role in sinus node activity, mutations in SCN5A have been linked to sick sinus syndrome, manifesting as sinus bradycardia, sinus arrest, sinoatrial block, or a combination of these conditions, which can progress to atrial inexcitability (atrial standstill). Loss-of-function SCN5A mutations result in reduced peak INa density, hyperpolarizing shifts in the voltage dependence of steady-state channel availability, and slow recovery from inactivation. These effects likely cause reduced automaticity, decreased excitability, and conduction slowing or block of impulses generated in the sinus node to the surrounding atrial tissue. Sinus node dysfunction can also manifest concomitantly with other phenotypes that are linked to SCN5A loss-of-function mutations such as Brugada syndrome and progressive cardiac conduction disorders.2,14
Familial Atrial Fibrillation
Loss-of-function mutations, gain-of-function mutations, and common polymorphisms on the SCN5A gene have been identified in some cases of atrial fibrillation (AF) occurring in young patients with structurally normal hearts. It is speculated that INa reduction may predispose to AF by slowing the electrical conduction velocity and thereby facilitating reentry. On the other hand, gain-of-function mutations can potentially predispose to AF by increasing atrial excitability. AF can occur in patients with other phenotypes of Na+ channelopathies, including LQT3, Brugada syndrome, dilated cardiomyopathy, and sinus node dysfunction. Furthermore, mutations in the SCN1B gene (encoding the β1 subunit of the Na+ channel) and the SCN2B gene (encoding the β2 subunit of the Na+ channel) have been identified in patients with AF, many of whom displayed ECG patterns suggestive of the Brugada syndrome.1,2
Dilated Cardiomyopathy
Some cases of familial dilated cardiomyopathy have been linked to SCN5A mutations. Dilated cardiomyopathy–linked SCN5A mutations cause diverse loss-of-function and gain-of-function changes in the gating properties, but how such changes evoke contractile dysfunction is not understood. It is speculated that SCN5A mutations disrupt the interactions between the mutant Na+ channels and intracellular (or extracellular) proteins that are essential for normal cardiomyocyte structure and architecture. Notably, dilated cardiomyopathy with SCN5A mutations display atrial or ventricular arrhythmias (including AF, VT, and VF), or both, as well as sinus node dysfunction, AV block, and intraventricular conduction delay.2
Acquired Diseases
In heart failure, peak INa is reduced (likely secondary to reduced SCN5A expression), whereas INaL is increased (likely because of increased phosphorylation of Na+ channels). Nav1.5 expression is reduced in the surviving myocytes in the border zone of the myocardial infarct (MI). Importantly, Na+ channel blockers can increase the risk for SCD in patients with ischemic heart disease, possibly by facilitating the initiation of reentrant excitation waves. Additionally, INaL increases during myocardial ischemia, explaining why INaL inhibition may be an effective therapy for chronic stable angina. Nav1.5 expression is reduced in response to persistent atrial tachyarrhythmias as part of the “electrical remodeling” process, leading to attenuation of INa.3
Potassium Channels
Structure and Physiology
Cardiac K+ channels are membrane-spanning proteins that allow the passive movement of K+ ions across the cell membrane along its electrochemical gradient. The ion-conducting or pore-forming subunit is generally referred to as the α subunit. The tripeptide sequence glycine-tyrosine-glycine GYG is common to the pore of all K+ channels and constitutes the signature motif for determining K+ ion selectivity. A gating mechanism controls switching between open-conducting and closed-nonconducting states.1,15
K+ channels represent the most diverse class of cardiac ion channels (Fig. 2-3). Cardiac K+ currents can be categorized as voltage-gated (Kv) and ligand-gated channels. In Kv channels, pore opening is coupled to the movement of a voltage sensor within the membrane electric field, and they include the rapidly activating and inactivating transient outward current (Ito); the ultrarapid (IKur), rapid (IKr), and slow (IKs) components of the delayed rectifier current; and the inward rectifier current (IK1). In contrast, pore opening in ligand-gated channels is coupled to the binding of an organic molecule, including channels activated by a decrease in the intracellular concentration of adenosine triphosphate (KATP) or activated by acetylcholine (KACh). Other classes of K+ channels respond to different stimuli, including changes in intracellular Ca2+ concentration and G proteins.15
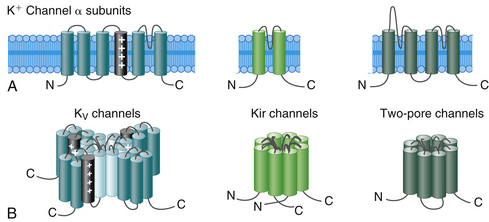
On the basis of the primary amino acid sequence of the α subunit, K+ channels have been classified into three major families (Table 2-2):
1. Channels containing six transmembrane segments and a single pore. This architecture is typical of Kv channels.
2. Channels containing two transmembrane segments (M1 and M2) and a pore. This architecture is typical of inward rectifier K+ (Kir) channels, including K1, KATP, and KACh channels. They conduct K+ currents more in the inward direction than the outward and play an important role in setting the resting potential close to the equilibrium potential for K+ and in repolarization. Kir channels form either homotetramers or heterotetramers.
3. Channels containing four transmembrane segments and two pores (K2P). These channels exist as homodimers or heterodimers. K2P currents display little time or voltage dependence. There are four classes of cardiac K2P: TASK, TWIK, TREK, and THIK.15,16
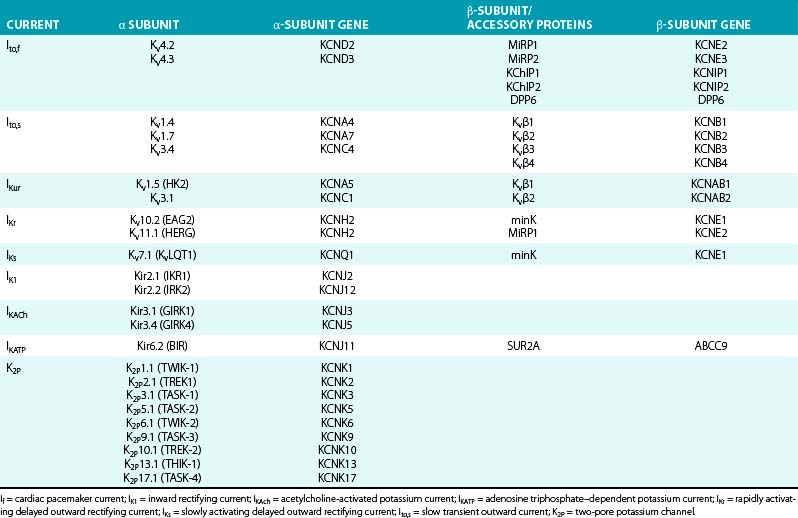
Each voltage-gated K+ channel (Kv family) is formed by the coassembly of 4 identical (homotetramers) or a combination of 4 different (from the same subfamily, heterotetramers) α subunits. A total of 38 genes has been cloned and assigned to 12 subfamilies of voltage-gated K channels (Kv1 to Kv12) on the basis of sequence similarities. Each α subunit contains one domain consisting of 6 membrane-spanning segments (S1 to S6), connected to each other by alternating intracellular and extracellular peptide loops (similar to 1 of the 4 domains of voltage-gated Na+ and Ca2+ channels), with both the amino terminus (N-terminus) and the C-terminus located on the intracellular side of the membrane. The central ion-conducting pore region is formed by the S5 and S6 segments and the S5-S6 linker (P segment); the S5-S6 linker is responsible for K+ ion selectivity. The S4 segment serves as the voltage sensor.1,15
The α subunits of Kv channels can generate voltage dependent K+ current when expressed in heterologous systems. However, the assembly of a functional tetramer can occur only in the presence of multiple auxiliary units (see Table 2-2). In many cases, auxiliary subunits coassociate with the α subunits and likely modulate cell surface expression, gating kinetics, and drug sensitivity of the α subunit complex. Most K+ channel β subunits assemble with α subunits and give rise to an α4β4 complex. K+ channel β subunits represent a diverse molecular group, which includes cytoplasmic proteins (Kvβ1 to Kvβ3, KChIP, and KChAP) that interact with the intracellular domains of Kv channels; single transmembrane spanning proteins (e.g., minK and minK-related proteins [MiRPs]) encoded by the KCNE gene family; and large ATP-binding cassette (ABC) transport-related proteins (e.g., the sulfonylurea receptors [SURs]).1,16
As with voltage-dependent Na+ and Ca2+ channels, Kv channels typically fluctuate among distinct conformational states because of molecular movements in response to voltage changes across the cell membrane (voltage-dependent gating). The Kv channel activates (opens) on membrane depolarization, thus allowing the rapid passage of K+ ions across the sarcolemma. After opening, the channel undergoes conformational transition in a time-dependent manner to a stable nonconducting (inactivated) state. Inactivated channels are incapable of reopening, even if the transmembrane voltage is favorable, unless they “recover” from inactivation (i.e., enter the closed state) on membrane repolarization. Closed channels are nonconducting but can be activated on membrane depolarization.17
Three mechanistically distinct types of Kv channel inactivation that are associated with distinct molecular domains have been identified: N-type, C-type, and U-type. N-type (“ball and chain”) inactivation involves physical occlusion of the intracellular mouth of the channel pore through binding of a small group of amino acids (“inactivation ball tethered to a chain”) at the extreme N-terminus. In contrast, C-type inactivation involves conformational changes in the external mouth of the pore. C-type inactivation exists in almost all K+ channels and may reflect a slow constriction of the pore. This inactivation process is thought to be voltage independent, coupled to channel opening, and is usually slower than N-type inactivation. Recovery from C-type inactivation is relatively slow and weakly voltage dependent. Importantly, the rate of C-type inactivation and recovery can be strongly influenced by other factors, such as N-type inactivation, drug binding, and changes in extracellular K+ concentration. These interactions render C-type inactivation an important biophysical process in regulating repetitive electrical activity and determining certain physiological properties such as refractoriness, drug binding, and sensitivity to extracellular K+.17
In addition to N-type and C-type inactivation, some Kv channels also show another type of inactivation (U-type), which exhibits a U-shaped voltage dependence with prolonged stimulation rates. Those channels appear to exhibit preferential inactivation from partially activated closed states, rapid and strongly voltage-dependent recovery from inactivation and, in some channel types, accelerated inactivation with elevation of extracellular K+. The exact conformational changes underlying U-type inactivation remain unclear. Importantly, there is extreme diversity in the kinetic and potentially molecular properties of Kv channel inactivation, particularly of C-type inactivation.17
Function
K+ channels are a diverse and ubiquitous group of membrane proteins that regulate K+ ion flow across the cell membrane on the electrochemical gradient and regulate the resting Em, the frequency of pacemaker cells, and the shape and duration of the cardiac action potential. Because the concentration of K+ ions outside the cell membrane is approximately 25-fold lower than that in the intracellular fluid, on activation, the opening of K+ channels generates an outward current resulting from the efflux of positively charged ions that offers a mechanism to counteract, dampen, or restrict the depolarization front (phases 1 through 4 of the action potential) triggered by an influx of cations (Na+ and Ca2+).1,3,15,16
The variation in the level of expression of K+ channels that participate in the genesis of the cardiac action potential explains the regional differences of the configuration and duration of cardiac action potentials from sinus node and atrial to ventricular myocytes and across the myocardial wall (endocardium, midmyocardium, and epicardium). Moreover, the expression and properties of K+ channels are not static; heart rate, neurohumoral state, pharmacological agents, cardiovascular diseases (cardiac hypertrophy and failure, MI) and arrhythmias (e.g., AF) can influence those properties, and they underlie the change in action potential configuration in response to variation in heart rate and various physiological and pathological conditions.1,15,16
Transient Outward Potassium Current (Ito)
Structure and Physiology
Cardiac Ito channels are macromolecular protein complexes, comprising four pore-forming Kv α subunits and a variety of Kv channel accessory (β) subunits (see Fig. 2-3). Ito is the sum of a voltage-dependent, Ca2+-independent K+ current (Ito1) and a Ca2+-activated Cl− or K+ current (Ito2). In human atrial and ventricular myocytes, the presence of Ito2 has not been clearly demonstrated.
Kv channels mediating Ito,s are formed by the coassembly of four α subunits from the Kv1.x subfamily (primarily Kv1.4, and possibly Kv1.7 and Kv3.4), whereas those mediating Ito,f are formed by the coassembly of four α subunits from the Kv4.x subfamily (primarily Kv4.3, and possibly Kv4.2) (see Table 2-2). Among the various accessory subunits identified, a crucial role has been definitively demonstrated only for KChIP2, and potentially for MiRP2.1,16,18
Function
The density of Ito varies across the myocardial wall and in different regions of the heart. In human ventricles, Ito densities are much higher in the epicardium and midmyocardium than in the endocardium. Furthermore, Ito,f and Ito,s are differentially expressed in the myocardium, thus contributing to regional heterogeneities in action potential waveforms. Ito,f is the principal subtype expressed in human atrium. The markedly higher densities of Ito,f, together with the expression of the ultrarapid delayed rectifier current, accelerate the early phase of repolarization and lead to lower plateau potentials and shorter action potentials in atrial as compared with ventricular cells.17,18
Although both Ito,f and Ito,s are expressed in the ventricle, Ito,f is more prominent in the epicardium and midmyocardium (putative M cells) than in the endocardium. These regional differences are responsible for the shorter duration and the prominent phase 1 notch and the “spike-and-dome” morphology of epicardial and midmyocardial compared with endocardial action potentials. A prominent Ito-mediated action potential notch in ventricular epicardium but not endocardium produces a transmural voltage gradient during early ventricular repolarization that registers as a J wave or J point elevation on the ECG.19 Ito densities are also reportedly higher in right than in left (midmyocardial and epicardial) ventricular myocytes, consistent with the more pronounced spike-and-dome morphology of right, compared with left, ventricular action potentials.1,3,17,18
Furthermore, variations in cardiac repolarization associated with Ito regional differences strongly influence intracellular Ca2+ transient by modulating Ca2+ entry via ICaL and Na+-Ca2+ exchange, thereby regulating excitation-contraction coupling and regional modulation of myocardial contractility and hence synchronizing the timing of force generation between different ventricular regions and enhancing mechanical efficiency.17
Regulation
Transient outward channels are subject to α- and β-adrenergic regulation. α-Adrenergic stimulation reduces Ito; concomitant β-adrenergic stimulation appears to counteract the α-adrenergic effect, at least in part. The effects of α- and β-adrenergic stimulation are exerted by phosphorylation of the Kv1.4, Kv4.2, and Kv4.3 α-subunits by PKA as well as PKC. Calmodulin-dependent kinase II, on the other hand, has been shown to be involved in enhancement of Ito. Adrenergic stimulation is also an important determinant of transient outward channel downregulation in cardiac disease. Chronic α-adrenergic stimulation and angiotensin II reduce Ito channel expression.20
KChIP2, when coexpressed with Kv4.3, increases surface channel density and current amplitude, slows channel inactivation, and markedly accelerates the recovery from inactivation. In the ventricle, KChIP2 mRNA is 25-fold more abundant in the epicardium than in the endocardium. This gradient parallels the gradient in Ito expression, whereas Kv4.3 mRNA is expressed at equal levels across the ventricular wall. Thus, transcriptional regulation of the KChIP2 gene (KChIP2) is the primary determinant of Ito expression in the ventricular wall.1,16
Observations suggest that MiRP2 is required for the physiological functioning of human Ito,f channels and that gain-of-function mutations in MiRP2 predispose to Brugada syndrome through augmentation of Ito,f.18
Ito is strongly rate dependent. Ito fails to recover from previous inactivation at very fast heart rates, which can be manifest as a decrease in the magnitude of the J wave on the surface ECG. Hence, abrupt changes in rate and pauses have important consequences for the early repolarization of the membrane.19
Ito can be enhanced by aging, low sympathetic activity, high parasympathetic activity, bradycardia, hypothermia, estrogen reduction, and drugs. Estrogen suppresses the expression of the Kv4.3 channel and results in reduced Ito and a shallow phase 1 notch.13
Phase 1 notch of the action potential modulates the kinetics of slower activating ion currents and consequently the later phases of the action potential. Initial enhancement of phase 1 notch promotes phase 2 dome and delays repolarization, presumably by delaying the peak of ICaL. However, further enhancement of phase 1 notch prevents the rising of phase 2 dome and abbreviates action potential duration, presumably by deactivation or voltage modulation that reduces ICaL. Thus, progressive deepening of phase 1 notch can cause initial enhancement followed by sudden disappearance of phase 2 dome and corresponding prolongation followed by abbreviation of action potential duration. On the other hand, modulators that decrease Ito lead to a shift of the plateau phase into the positive range of potentials, thus increasing the activation of the delayed rectifier currents, promoting faster repolarization, and reducing the electrochemical driving force for Ca2+ and hence ICaL. Phase 1 notch also affects the function of the Na+-Ca2+ exchanger and subsequently intracellular Ca2+ handling and Na+ channel function.1,16
Pharmacology
Quinidine, 4-aminopyridine, flecainide, and propafenone produce an open channel blockade and accelerate Ito inactivation. Quinidine, but not flecainide or propafenone, produces a frequency-dependent block of Ito that results from a slow rate of drug dissociation from the channel. Quinidine has relatively strong Ito blocking effect, whereas flecainide mildly blocks Ito.18,20
Ito blockers can potentially prolong the action potential duration in the atrial and in ischemic ventricular myocardium. However, because the net effects of Ito blockade on repolarization depend on secondary changes in other currents, the reduction of Ito density can result in a shortening of the ventricular action potential. Moreover, heterogeneous ventricular distribution of Ito can cause marked dispersion of repolarization across the ventricular wall that, when accompanied by prominent conduction delays related to Na+ channel blockade, results in extrasystolic activity through a phase 2 reentrant mechanisms.18,20
Currently, a cardioselective and channel-specific Ito opener or blocker is not available for clinical use. Development of an Ito-selective drug is expected to be beneficial in patients with primary abnormality in the Ito or in other channels, such as the Brugada syndrome, in which heterogeneity in the expression of Ito between epicardium and endocardium in the right ventricle (RV) results in the substrate responsible for reentry and ventricular arrhythmias.18,20
Inherited Channelopathies
To date, only mutations in KCNE3 (MiRP2) are linked to inherited arrhythmia. KCNE3 mutations were identified in five related patients with Brugada syndrome. When expressed with Kv4.3, the mutation increased Ito,f density.3
Another KCNE3 mutation was identified in one patient with familial AF. The mutation was found to increase Ito,f and was postulated to cause AF by shortening action potential duration and facilitating atrial reentrant excitation waves.3
A genome wide haplotype-sharing study associated a haplotype on chromosome 7, harboring DPP6, with idiopathic VF in three distantly related families. Overexpression of DPP6, which encodes dipeptidyl-peptidase 6, a putative component of the Ito channel complex, was proposed as the likely pathogenetic mechanism. DPP6 significantly alters the inactivation kinetics of both Kv4.2 and Kv4.3 and promotes expression of these α subunits in the cell membrane.3,21
Importantly, the normally functioning Ito channels play an important role in the electrophysiological consequences of the ionic current abnormalities in the Brugada and J wave syndromes. Heterogeneity in the distribution of Ito channels across the myocardial wall, being more prominent in ventricular epicardium than endocardium, and particularly in the RV, results in the shorter duration and the prominent phase 1 notch and the spike-and-dome morphology of the epicardial action potential as compared with the endocardium. The resultant transmural voltage gradient during the early phases (phases 1 and 2) of the action potential is thought to be responsible for the inscription of the J wave on the surface ECG.18,19 An increase in net repolarizing current, secondary to either a decrease in the inward currents (INa and ICaL) or an increase in the outward K+ currents (Ito, IKr, IKs, IKAch, IKATP), or both, can accentuate the action potential notch and lead to augmentation of the J wave or the appearance of ST segment elevation on the surface ECG. An outward shift of currents that extends beyond the action potential notch not only can accentuate the J wave but also can lead to partial or complete loss of the dome of the action potential, thus leading to a protracted transmural voltage gradient that manifests as greater ST segment elevation and gives rise to J wave syndromes. The type of the ion current affected and its regional distribution in the ventricles determine the particular phenotype (including the Brugada syndrome, early repolarization syndrome, hypothermia-induced ST segment elevation, and MI-induced ST segment elevation).18,19,22 The degree of accentuation of the action potential notch leading to loss of the dome depends on the magnitude of Ito. These changes are more prominent in regions of the myocardium exhibiting a relatively large Ito, such as the RV epicardium; this explains the appearance of coved ST segment elevation, characteristic of Brugada syndrome, in the right precordial ECG leads.19,20
In this context, factors that influence the kinetics of Ito or the other repolarization currents can modify the manifestation of the J wave on the ECG. Na+ channel blockers (procainamide, pilsicainide, propafenone, flecainide, and disopyramide), which reduce the inward INa, can accentuate the J wave and ST segment elevation in patients with concealed J wave syndromes. Quinidine, which inhibits both Ito and INa, reduces the magnitude of the J wave and normalizes ST segment elevation. Additionally, acceleration of the heart rate, which is associated with reduction of Ito (because of slow recovery of Ito from inactivation), results in a decrease in the magnitude of the J wave. Male predominance can potentially result from larger epicardial Ito density versus Ito in women.19,22
The increased transmural heterogeneity of ventricular repolarization (i.e., dispersion of repolarization between epicardium and endocardium), which is responsible for J point elevation and early repolarization pattern on the surface ECG, is also responsible for the increased vulnerability to ventricular tachyarrhythmias. A significant outward shift in current can cause partial or complete loss of the dome of the action potential in regions where Ito is prominent (epicardium), with the consequent loss of activation of ICaL. The dome of the action potential then can propagate from regions where it is preserved (midmyocardium endocardium) to regions where it is lost (epicardium), thus giving rise to phase 2 reentry, which can generate premature ventricular complexes that in turn can initiate polymorphic VT or VF.19,20,22,23
Acquired Diseases
An alteration in the expression and distribution of Ito is observed in various pathophysiological conditions. Adrenergic effects seem to be involved in at least some of these Ito-regulating processes during heart disease.20
In general, myocardial ischemia, MI, dilated cardiomyopathy, and end-stage heart failure cause downregulation of Ito. In fact, Ito downregulation is the most consistent ionic current change in the failing heart. The reduction in Ito results in attenuation of early repolarization (phase 1) and affects the level of plateau (phase 2) of the action potential and other currents involved in delayed repolarization (phase 3), with resulting prolongation and increased heterogeneity of action potential duration. The prominent epicardial Ito contributes to the selective electrical depression of the epicardium. This process leads to the development of a marked dispersion of repolarization between normal and abnormal epicardium and between epicardium and endocardium, which provides the substrate for reentrant arrhythmias and may underlie the increased predisposition to ventricular arrhythmias and SCD in patients with heart failure and ischemic heart disease.18,20,24 Additionally, downregulation of Ito in advanced heart failure likely slows the time course of force generation, thereby contributing to reduced myocardial performance.
On the other hand, compensatory ventricular hypertrophy preceding heart failure is associated with an upregulation of Ito. The prolongation of the action potential, concomitant with an increase in Ito, presumably results from the more negative level of the plateau with less ICaL inactivation and probably less delayed rectifier activation. In contrast, progression of hypertrophy to heart failure is associated with a clear reduction in Ito.20
Chronic AF reduces Ito density and Kv4.3 mRNA levels. Hypothyroidism reduces the expression of KCND2 (Kv4.2) genes. Additionally, Ito may also be reduced and contribute to QT interval prolongation in diabetes. Importantly, with some delay, insulin therapy partially restores Ito, maybe by enhancing Kv4.3 expression.3
Ultrarapidly Activating Delayed Outward Rectifying Current (IKur)
Structure and Physiology
The ion-conducting pore of IKur channels is formed by four Kv1.5 α subunits, whereas the ancillary β subunits Kvβ1.2, Kvβ1.3, and Kvβ2.1 control channel trafficking and plasma membrane integration as well as activation and inactivation kinetics.3,25
IKur activates rapidly on depolarization in the plateau range and displays outward rectification, but it inactivates very slowly during the time course of the action potential. Inactivation accelerates when Kv1.5 is coexpressed with its β subunits.3
Function
IKur is detected only in human atria and not in the ventricles, so that it is the predominant delayed rectifier current responsible for human atrial repolarization and is a basis for the much shorter duration of the action potential in the atrium.1,3
Regulation
β-Adrenergic stimulation enhances IKur, whereas α-adrenergic stimulation inhibits it, effects likely mediated by PKA and PKC, respectively. Membrane depolarization and elevated extracellular K+ concentrations reduce Kv1.5 expression. Additionally, cAMP, mechanical stretch, hyperthyroidism, and dexamethasone increase Kv1.5 expression, whereas extracellular acidosis, phenylephrine, and hypothyroidism decrease it.1,3
Pharmacology
IKur is relatively insensitive to class III antiarrhythmics of the methane-sulfonanilide group, but it is highly sensitive to 4-aminopyridine. Selective inhibition of IKur by 4-aminopyridine prolongs the human atrial action potential duration.16
IKur is a promising target for the development of new, safer antiarrhythmic drugs to prevent AF or atrial flutter, or both, without a risk of ventricular proarrhythmia. Because IKur is atrium specific, a drug specifically targeting Kv1.5 channels would be expected to terminate AF by preventing reentry through atrial action potential prolongation. The drug vernakalant is an IKur/INa channel blocker and is undergoing review by the U.S. Food and Drug Administration for the acute termination of AF. However, because Kv1.5 is downregulated in AF, the beneficial effect of IKur block becomes less certain. Furthermore, because Kv1.5 is also expressed in other organs (e.g., brain), discovery of drugs that selectively inhibit atrial Kv1.5 channels remains necessary.1,3,16
Physiologically, rapid activation of IKur in the positive potential range following the action potential upstroke can offset depolarizing ICaL and hence lead to the less positive plateau phase in atrial compared with ventricular cardiomyocytes. Conversely, block of IKur produces a more pronounced spike-and-dome configuration and therefore shifts the potential into a more positive range in which ICaL activation enhances systolic Ca2+ influx during a free-running action potential. Such an indirect effect on ICaL should be shared by all IKur blockers and is expected to result in a positive atrial inotropic effect.25






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


