Essentials of Diagnosis
- Recurrent attacks of oral aphthous ulcers, genital ulcers, uveitis, and skin lesions.
- Onset usually in young adults, aged 25–35 years.
- Prevalent in parts of Asia and Europe; rare in North America.
- Blindness, central nervous system disease, and large-vessel events are the most serious complications.
- Glucocorticoids, immunosuppressive drugs, or both are required for severe disease.
General Considerations
Behçet disease, a form of vasculitis of unknown cause, is named for the Turkish dermatologist who in 1937 described the syndrome as a triad of recurrent oral aphthous ulcers, genital ulcers, and ocular inflammation. Although these features are often the most salient, Behçet disease can cause inflammation in almost any organ. Indeed, involvement of the central nervous system, gastrointestinal tract, and large vessels can be life-threatening. Except for eye disease, most of the manifestations of Behçet disease do not persist chronically but recur in attacks, which usually become less frequent over time. Disability stems most often from ocular inflammation, which causes blindness, and less often from central nervous system disease. Mortality results chiefly from major vascular events, including thrombosis, aneurysm, and rupture of large vessels.
Epidemiology
One of the most striking features of Behçet disease is how common it is in countries along the ancient Silk Road and how rarely it develops elsewhere. Most prevalent in Turkey (up to nearly 400 cases per 100,000 people), Behçet disease also occurs frequently in Iran, Saudi Arabia, Greece, Japan, Korea, and China. In contrast, Behçet disease rarely develops in Western countries such as the United States, where the disease affects about 1 of every 170,000 people.
Etiology & Pathogenesis
Although the cause of Behçet disease is unknown, the distinct geographic clustering of cases suggests the importance of environment, genes, or both. Genetic studies have revealed a strikingly high prevalence of the HLA-B51 allele in patients living along the Silk Road, reaching nearly 80% of Asian patients. However, this allele is not associated with Behçet disease in Western countries.
Much of the damage in Behçet disease results from blood vessel inflammation, justifying the disease’s classification as a form of vasculitis. Although Behçet disease typically affects the small- and medium-sized vessels, it is one of the rare forms of vasculitis capable of also affecting large arteries. Arterial inflammation can lead to occlusion, aneurysm, or rupture. Behçet disease joins granulomatosis with polyangiitis (formerly Wegener granulomatosis) and Buerger disease in being a form of vasculitis that has a predilection for involving veins and causing venous thrombosis.
Clinical Findings
Oral ulceration is the hallmark of the disease, tends to be the earliest manifestation, and is required for the diagnosis of Behçet disease (Table 38–1). Oral ulcers are painful, shallow or deep, round or oval, with a white or yellow base and red halo (Figure 38–1). They vary in size from 1–20 mm. The ulcers most frequently affect the buccal mucosa, tongue, lips, gingivae, palate, tonsils, uvula, or pharynx. During an attack, patients usually have two to five lesions, but some patients may have a single ulcer or too many to count. The aphthae may be so painful that the patient has trouble eating or drinking. Usually the aphthous lesions heal without scarring over 10–20 days.
| Feature | Frequency (%) |
|---|---|
| Oral ulcers | 100 |
| Genital ulcers | 75 |
| Skin lesions | 60–90 |
| Arthritis | 50 |
| Gastrointestinal disease | 25 |
| Thrombophlebitis | 20 |
| Central nervous system disease | 10–20 |
| Epididymitis | 5 |
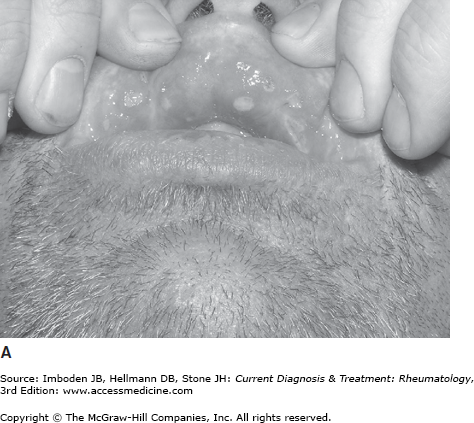
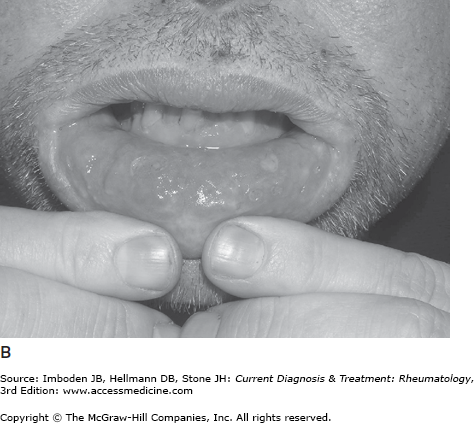
Genital aphthae occur slightly less often than oral ulceration (Table 38–1). However, genital ulcers tend to be larger and deeper, and often heal with scarring. In men, the ulcers develop most commonly on the scrotum and less commonly on the shaft of the penis, and in women ulcers affect the vagina and vulva. Genital lesions in men are often associated with epididymitis.
Cutaneous manifestations of Behçet disease, which develop in 60–90% of patients, are protean. Erythema nodosum occurs most commonly, especially in women. Erythema nodosum in Behçet disease tends to ulcerate and heal with scarring and hyperpigmentation, compared with erythema nodosum associated with sarcoidosis and inflammatory bowel disease, which does not ulcerate and heals without scarring. In men, pseudofolliculitis and acneiform nodules develop frequently over the neck and face. Pathergy—the phenomenon of developing an aseptic nodule or ulcer larger than 2 mm in diameter 24–48 hours following a sterile needle prick to the forearm—occurs frequently in Japanese and Turks but in only approximately one third of Americans with Behçet disease. Migratory thrombophlebitis also commonly occurs in Behçet disease.
Ocular inflammation, one of the hallmark manifestations of Behçet disease, tends to occur early in the course. Recurrent or persistent ocular inflammation frequently leads to visual loss, making eye inflammation one of the most common causes of disability in Behçet disease. Behçet disease is one of the few autoimmune diseases that can cause both anterior and posterior uveitis. Anterior uveitis typically presents with a red eye, intense photophobia, and blurred vision. The anterior uveitis may be so intense that a grossly visible layer of pus in the anterior chamber (hypopyon) develops. The posterior uveitis and vasculitis of the carotid and retina occur less commonly but pose a greater threat to vision.
Peripheral arthritis or spondylitis develops in approximately half of patients with Behçet disease. The peripheral arthritis may be monarticular or polyarticular, while the spondylitis usually presents as sacroiliitis (with low back or buttock pain). The peripheral arthritis is usually not deforming.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


