Fig. 2.1
Schematic representation of the dystrophin-glycoprotein complex and dystroglycan
Mutations in components of the DGC, such as dystrophin and sarcoglycans, were identified to be associated with Duchenne/Becker and limb-girdle type-muscular dystrophies in the late 1980s to the mid-1990s. Although mutations in the DAG1 gene, which encodes DG, were not identified until 2011 [3], there was a unique route leading to the establishment of the disease entity called α-DGpathy. In 2001, Hayashi and colleagues reported that the reactivity of the monoclonal antibody IIH6 that recognizes a portion of the O-glycan structure of α-DG is dramatically reduced in patients with Fukuyama congenital muscular dystrophy (FCMD) [4]. Subsequently, Campbell and colleagues confirmed that the α-DG proteins from patients with FCMD, Walker-Warburg syndrome (WWS), and muscle-eye-brain disease (MEB) were abnormally glycosylated and had lost their ligand-binding activities [5]. Abnormal α-DG glycosylation was subsequently also found in patients with congenital muscular dystrophy (CMD) and limb-girdle muscular dystrophy (LGMD) [6–8]. In this way, the concept of α-DGpathy was established [9, 10]. Currently, there are around 15 known α-DGpathy genes, all of which are involved either directly or indirectly in the glycosylation of α-DG (Table 2.1). However, some unidentified causative genes for α-DGpathy could possibly be identified in the near future.
Table 2.1
Dystroglycanopathy genes and their functions
Dystroglycanopathy genes | Gene functions | MDDG number |
|---|---|---|
POMT1 | O-mannosyltransferase (POMT1/POMT2 complex) | 1 |
POMT2 | O-mannosyltransferase (POMT1/POMT2 complex) | 2 |
POMGNT1 | Protein O-mannose β1,2-GlcNAc transferase | 3 |
FKTN | Unknown, likely involved in post-phosphoryl modification | 4 |
FKRP | Unknown, likely involved in post-phosphoryl modification | 5 |
LARGE | Synthesis of Xyl-GlcA repeating units | 6 |
ISPD | Unknown | 7 |
GTDC2/POMGNT2 | Protein O-mannose β1,4-GlcNAc transferase | 8 |
DAG1 | Dystroglycan, reduced interaction with LARGE | 9 |
TMEM5 | Unknown | 10 |
B3GALNT2 | β1,3-GalNAc transferase | 11 |
SGK196/POMK | O-Man kinase | 12 |
B3GNT1(B4GAT1) | β1,4-GlcA transferase | 13 |
GMPPB | GDP-mannose pyrophosphorylase B | 14 |
DPM1 | Dol-P-Man synthesis | – |
DPM2 | Dol-P-Man synthesis | – |
DPM3 | Dol-P-Man synthesis | – |
DOLK | Dolichol phosphate synthesis | – |
FCMD is an autosomal recessive muscular dystrophy that is the most common CMD in Japan. FCMD is also the most common childhood muscular dystrophy, next to Duchenne muscular dystrophy (DMD), in Japan [11]. The incidence of FCMD is around 3 per 100,000 persons, and one in about 90 persons is expected to be a heterozygous carrier. FCMD is characterized by CMD in combination with abnormalities of the central nervous system (brain malformations characterized by micropolygyria of the cerebrum and cerebellum and type II lissencephaly) and the eye (severe myopia, cataracts, optic nerve hypoplasia, retinal dysplasia, etc.) [12]. Severe mental retardation is observed in all cases. Most patients are never able to work, and they die by the age of 20 years.
MEB is a severe autosomal recessive disease characterized by CMD, ocular abnormalities, and brain malformations and is seen mainly in Finland. As with FCMD, hypoglycosylation of α-DG is commonly seen in MEB patients [5, 13]. WWS is the most severe form of α-DGpathy, with most patients living only a year or less. Similar to FCMD and MEB, this disease is characterized by CMD, structural brain defects, and eye malformations. WWS patients show a high degree of genetic heterogeneity, and several genes have been implicated in this disease.
All of these disorders are biochemically characterized by abnormal glycosylation of α-DG and are clinically associated with severe CMD and brain abnormalities. On the other hand, a considerable number of patients who show only mild muscular dystrophy without brain malformations have been also reported [14, 15]. A number of studies on large cohorts of α-DGpathy patients confirmed that there is a wide clinical spectrum, and there is no clear genotype-phenotype correlation [16, 17]. It is thought that effects of mutation on the gene function, rather than types of disease-causing gene, affect disease severity. The most severe end of the spectrum is characterized by CMD with extensive structural abnormalities in the brain and eyes, which usually results in early infantile death. Patients at the mildest end of the spectrum may present in adult life with LGMD, without the involvement of the brain or eyes.
In 2007, Muntoni and colleagues proposed a clinical classification system comprising seven broad phenotypic categories [16]. They first categorized α-DGpathy into four groups: (a) WWS/WWS-like, (b) MEB/FCMD-like, (c) CMD, and (d) LGMD. Categories (c) and (d) were further classified into subgroups: (c-1) CMD with mental retardation and cerebellar involvement as the only structural brain abnormality, (c-2) CMD with mental retardation and a structurally normal brain, (c-3) CMD with no abnormal cognitive development, (d-1) LGMD with mental retardation, and (d-2) LGMD without mental retardation. Later, new Online Mendelian Inheritance in Man (OMIM) entries created a simplified classification scheme for α-DGpathy (MDDG; muscular dystrophy dystroglycanopathy) by combining three broad phenotypic groups and gene defects. First, α-DGpathy was divided into three groups: (A) CMD with brain/eye abnormalities, (B) CMD with milder brain structural abnormalities, and (C) LGMD. Second, the causative gene was indicated numerically (e.g., POMT1 is “1” and fukutin is “4”). From this classification, typical FCMD is named “MDDG type A4.” For more information, please refer to Godfrey et al. [17].
2.2 Sugar Chain Structure and Modification Mechanisms of α-DG
Glycosylation is a posttranslational modification that attaches sugar moieties to proteins or lipids and plays crucial roles in a wide range of biological processes. Glycosylation of α-DG also plays a central role in the functional maturation of α-DG as well as in the pathogenesis of α-DGpathy. In this section, the structure of sugar chain and mechanisms by which it is modified are reviewed.
DG consists of α and β subunits, both of which are expressed from a single mRNA and cleaved into α-DG and β-DG during posttranslational modifications [18]. α-DG is a highly glycosylated extracellular protein that functions as a ligand-binding subunit for several ligand proteins that commonly contain laminin G (LG) domain-like modules. β-DG is a transmembrane subunit that anchors α-DG on the cell surface and binds to dystrophin inside the cell. α-DG consists of three domains, two globular domains at the N- and C-termini and an intervening mucin-like domain (Fig. 2.2). The mucin-like domain contains more than 40 Ser/Thr residues that form O-glycan clusters. The N-terminal globular domain is essential for O-glycosylation to proceed in the mucin-like domain. However, the N-terminal domain is cleaved and shed from the α-DG core portion after the completion of glycosylation and is therefore not present in mature α-DG [19]. The significance of this cleavage is not understood. Because glycosylation, which is necessary for ligand binding, takes place a few amino acids downstream of the cleavage site, it may provide a stereochemically efficient structure for interaction with the ligands [20]. Among heterogeneous groups of O-linked sugar chains on the mucin-like domain of α-DG, a unique O-mannose (O-Man) type sugar chain is important for the ligand-binding activity.
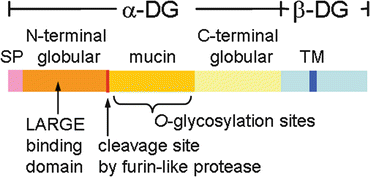
Fig. 2.2
Schematic representation of dystroglycan. The domain structure of dystroglycan. SP signal peptide, TM transmembrane
O-Mannosyl glycan structures of α-DG that are currently known are shown in Fig. 2.3. Core M1 (Siaα2–3Galβ1–4GlcNAcβ1–2Man), which was the first O-Man type glycan found in mammals, was originally identified from the bovine peripheral nerve by Endo and colleagues [21]. The structure of Core M2 is currently presumed based on the finding that N-acetylglucosaminyltransferase IX (GnT-IX) can transfer GlcNAc to O-Man [22]. Core M3 (GalNAcβ1–3GlcNAcβ1–4Man) was identified in recombinant α-DG proteins expressed in HEK293 cells [23]. In 2010, Campbell and colleagues reported the presence of a novel moiety that is modified on O-Man via a phosphodiester linkage and proposed that this moiety forms a ligand-binding domain [23]. Because its complete structure has not yet been elucidated, this structure has been tentatively termed as the “post-phosphoryl moiety” or “post-phosphoryl modification.” It is likely that this post-phosphoryl moiety contains repeat units consisting of xylose (Xyl) and glucuronic acid (GlcA) and that these units are assembled through the enzymatic activity of LARGE (like-acetylglucosaminyltransferase) [24]. Enzymatically synthesized Xyl-GlcA repeats bind to laminin, and their ligand-binding capacity is associated with the length of the repeats [25]. Overexpression of LARGE produces hyperglycosylated α-DG that has increased ligand-binding activity [19, 26]. This activity is likely due to the formation of longer and/or more repeating units. Taken together, it appears that the repeating Xyl-GlcA units function as the ligand-binding epitopes of α-DG. Details of the linker structure between the phosphorylated Man (Man-P) and the Xyl-GlcA repeats remain unknown. Currently, more than 15 genes have been identified in which mutations cause abnormal glycosylation of α-DG resulting in human disease (Table 2.1). The enzymatic or putative functions of these gene products are described in the following paragraphs.
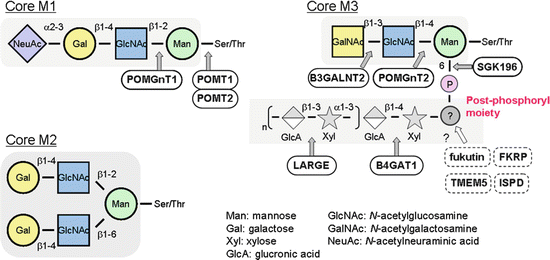
Fig. 2.3
Sugar chain structures of α-dystroglycan. Unique O-mannose glycans of α-dystroglycan and enzymes involved in their synthesis are illustrated
Protein O-mannosyltransferase (POMT) 1 and POMT2 were originally identified as the genes responsible for WWS [27, 28]. POMT1 and POMT2 localize to the endoplasmic reticulum (ER) and form a POMT1/POMT2 heterocomplex [29]. This POMT complex catalyzes the transfer of a mannosyl residue from dolicholphosphate mannose (Dol-P-Man) to the Ser and Thr residues of α-DG.
Protein O-linked mannose β1,2-N-acetylglucosaminyltransferase 1 (POMGNT1) was cloned from a cDNA sequence that is homologous to N-acetylglucosaminyltransferase I (GnT-I, encoded by MGAT1) and was identified as a causative gene for MEB [30]. POMGnT1 is a glycosyltransferase that catalyzes the formation of the GlcNAcβ1–2Man linkage by transferring GlcNAc from UDP-GlcNAc to O-Man. The enzymatic removal of NeuAc, Gal, and GlcNAc from Core M1 does not affect the ligand-binding activity of α-DG [31]. This suggests that Core M1 may not directly serve as a ligand-binding domain. However, in POMGnT1-deficient mice, and fibroblasts from MEB patients, the post-phosphoryl modification is partially disrupted, raising the possibility that the structure of Core M1 may influence the biosynthesis of the Core M3/post-phosphoryl moiety [23, 32].
Glycosyltransferase-like domain containing 2 (GTDC2, POMGNT2), β1,3-N-acetylgalactosaminyltransferase 2 (B3GALNT2), and SGK196 (POMK) are newly discovered α-DGpathy genes that are involved in the synthesis of Core M3 [33–36]. GTDC2 (alternatively called AGO61) is localized in the ER and catalyzes the formation of the GlcNAcβ1–4Man linkage by the transfer of GlcNAc from UDP-GlcNAc to the O-mannosyl peptide derived from α-DG. B3GALNT2 is also localized in the ER and catalyzes the formation of the GalNAcβ1–3GlcNAc linkage by transferring GalNAc from UDP-GalNAc. SGK196 catalyzes the addition of a phosphate group from ATP to position 6 on the O-Man residues in the Core M3 structure. Mutations in any molecule that participates in the synthesis of Core M3 result in α-DGpathy. Therefore, highly ordered and sequential actions of the POMT complex, GTDC2, B3GALNT2, and SGK196 are required for subsequent post-phosphoryl modifications. It is not known whether Core M1 and Core M3 can be simultaneously modified on the same O-Man residues. In addition, because defects in either Core M1 or Core M3 synthesis result in the loss of the ligand-binding activities of α-DG, it is necessary to uncover the nature of their relationship in the biosynthetic pathway and/or in the state of ligand binding.
LARGE was originally discovered as a gene that was defective in meningiomas [37] and was later shown to be causative for spontaneous muscular dystrophy in Large myd mice [38] and CMD 1D (MDC1D) in humans [8]. LARGE possesses both xylosyltransferase and glucuronyltransferase activities that produce repeating units of [−3Xylα1–3GlcAβ1–] using UDP-GlcA and UDP-Xyl as donor substrates [24]. In addition to the enzymatic activities of LARGE, a physical interaction between LARGE and the α-DG N-terminal domain is required for the functional maturation of α-DG [19]. Thus, the N-terminal domain serves as a recognition motif for LARGE, thereby providing substrate specificity for this unique modification. As mentioned previously, a missense mutation in the DAG1 gene was identified in an α-DGpathy patient. The mutation was located within the N-terminal globular domain of α-DG, which leads to reduced interactions with LARGE and consequently impairs the formation of Xyl-GlcA repeating units [3].
Mutations in B3GNT1 were identified in WWS patients [39], and its gene product was thought to be required for LARGE-dependent glycosylation of α-DG [40]. Recently, it has been revealed that B3GNT1 encodes for a β1,4-glucuronyltransferase to form a GlcAβ1–4Xyl disaccharide and thus proposed to rename the enzyme B4GAT1 [41, 42]. The GlcAβ1–4Xyl acts as an acceptor primer that can be elongated by LARGE (Fig. 2.3).
Fukutin (FKTN) was identified as the gene responsible for FCMD [43]. Fukutin-related protein (FKRP) was identified based on sequence homology to fukutin [6]. Both fukutin and FKRP are localized in the Golgi apparatus and are involved in the post-phosphoryl modifications of α-DG [23, 32], although their exact functions remain unknown. It is expected that mutations in fukutin or FKRP reduce the activities of these proteins in α-DG glycosylation. Interestingly, however, several disease-causing point mutations cause misfolding of the fukutin protein resulting in changing its cellular localization from the Golgi apparatus to the ER, which also triggers abnormal glycosylation of α-DG [44]. A database retrieval study showed that there is a region in the fukutin protein that is homologous to bacterial proteins involved in polysaccharide/phosphorylcholine modifications and to a yeast protein involved in the mannosyl phosphorylation of oligosaccharides [45]. Another report proposed that fukutin and FKRP belong to the nucleotidyltransferase (NTase) fold protein superfamily [46].
TMEM5 is a newly discovered α-DGpathy gene that encodes transmembrane protein 5 [35, 47]. The TMEM5 protein contains the exostosin family domain found in EXT1, a glycosyltransferase required for the biosynthesis of heparan sulfate. The exact function of TMEM5 is unknown, but it is expected to be a glycosyltransferase.
The isoprenoid synthase domain-containing protein (ISPD) belongs to the 4-diphosphocytidyl-2C-methyl-D-erythritol (CDP-ME) synthase family, and mutations in the ISPD gene were found in WWS patients [48, 49]. ISPD contributes to the synthesis of isoprenoid precursors in the methylerythritol phosphate (MEP) pathway in Escherichia coli. However, it is postulated that the MEP pathway is absent in mammals. Therefore, specific roles for ISPD, especially in relation to α-DG glycosylation, are still unknown in humans. It has been proposed that the function of ISPD is crucial for efficient POMT-dependent O-mannosylation or synthesis of a novel nucleotide sugar that is required for α-DG glycosylation.
Defects in the synthesis pathway of Dol-P-Man are associated with α-DGpathy. Dol-P-Man is a Man donor in reactions that include N-glycosylation, O-mannosylation, and glycophosphatidylinositol (GPI) anchor biosynthesis in the ER lumen. Dol-P-Man is synthesized from GDP-Man and dolichol phosphate by the DPM complex, which consists of the catalytic component DPM1 and the ER-localized transmembrane proteins, DPM2 and DPM3. Mutations in DPM1, DPM2, or DPM3 have been identified in patients that show α-DGpathy with a type I congenital disorder of glycosylation (CDG) [50–52]. CDG is a heterogeneous group of rare genetic disorders that were originally defined as defects in the N-glycosylation process and are now reclassified to include O-linked and lipid glycosylation defects. In addition, mutations in the gene for dolichol kinase (DOLK) that is responsible for the formation of dolichol phosphate were discovered in patients with CDG and dilated cardiomyopathy [53]. Heart tissues from these patients showed abnormal O-mannosylation of α-DG. Mutations in the GDP-Man pyrophosphorylase B (GMPPB) gene that is responsible for the formation of GDP-Man were also reported in α-DGpathy patients [54]. Presumably, mutations in GMPPB result in a decrease in GDP-Man and consequently Dol-P-Man levels. Taken together, defects in the Dol-P-Man synthesis pathway are expected to perturb O-mannosylation of α-DG. However, it is also expected that defects in the synthesis of Dol-P-Man would affect O-mannosylation, N-glycosylation, and GPI anchor formation of other proteins. In fact, defects in N-glycosylation were found in patients with DPM mutations [50–52]. It is possible that defects in reactions using Dol-P-Man, other than O-mannosylation of α-DG, may also contribute to the pathology of these diseases. Thus, it remains controversial whether or not gene mutations in the Dol-P-Man biosynthesis pathway can be attributed to a cause of α-DGpathy.
2.3 Pathophysiological Function of DG
Several extracellular matrix and synaptic proteins such as laminins [55–58], agrin [59, 60], perlecan [61], neurexin [62], pikachurin [63], and slit [64] are ligands of α-DG. All of these ligand proteins commonly contain LG domain-like modules that function as binding sites for α-DG. Because O-mannosylations and post-phosphoryl modifications are required for the ligand-binding activities of α-DG, the DG-ligand interactions are dramatically reduced in patients with α-DGpathy.
Laminin is a major component of the basement membrane and is composed of three subunits (α, β, and γ) [65]. The expression pattern of each laminin isoform (currently, more than 15 different isoforms are known) differs among different tissues and cells. In the skeletal muscle basement membrane, the laminin α2 subunit of the main laminin isoform (laminin-211, composed of α2, β1, and γ1 subunits) interacts with α-DG on the surface of muscle cells. This interaction establishes a physical link between the basement and plasma membranes.
Agrin is a heparan sulfate proteoglycan and agrin-DG interactions play important roles in the structural maintenance and stabilization of neuromuscular junctions [66]. Perlecan is also a heparan sulfate proteoglycan and one of the major components of basement membranes. The relatively high binding affinities in perlecan-DG interactions possibly indicate that there are specific roles for these interactions [56].
Neurexins are transmembrane proteins that are expressed in the nervous system. There are two families of neurexins, α- and β-neurexins, and both can bind to α-DG. The significance of the neurexin-DG interaction is unclear, but it may be involved in the formation and/or maintenance of synapses because neurexins are located on the presynaptic membranes, and α-DG is expressed on the postsynaptic membranes [67].
Pikachurin is expressed in the synaptic clefts of the photoreceptor ribbon synapses. Interactions between pikachurin and DG play important roles in the maintenance of retinal synapse structures, synaptic signal transmissions, and visual functions [63, 68]. Slit is a protein widely referred to as a repulsive axon guidance cue, and Robo is its known transmembrane receptor. Slit-DG interactions are required for the proper localization of Slit, suggesting a new function for α-DG as an extracellular scaffold for controlling axon guidance [64].
Several mouse models for α-DGpathy have been established, and studies using these models have helped reveal pathophysiological roles of α-DG glycosylation (Table 2.2). Targeted disruptions of α-DGpathy genes resulted in embryonic lethality (POMT1, fukutin) [69, 70] or early postnatal lethality (GTDC2) [71]. POMGnT1 knockout (KO) mice are viable but show variable life spans [72, 73]. To bypass these issues, chimeric mice, knock-in (KI) mice, or conditional KO (cKO) mice have been generated. Fukutin-deficient chimeric mice, generated using embryonic stem cells targeted for both Fktn alleles, develop severe muscular dystrophy and display brain and eye anomalies [74]. Fukutin KI mice carrying the most prevalent FCMD mutation (retrotransposal insertion) confirmed that the retrotransposal insertion leads to abnormal post-phosphoryl modification of α-DG [32, 75]. Skeletal muscle-selective fukutin-cKO mice recapitulate features of α-DGpathy [76, 77]. Ablation of the putative functional domain from Fkrp (E310 to the TGA stop) results in embryonic lethality; the mutant homozygous embryos die before reaching embryonic day 12.5 [78]. Homozygous KI mice carrying the missense P448L mutation develop muscular dystrophy as well as a wide range of structural abnormalities in the central nervous system that are characteristic of neuronal migration defects [78]. Abnormal phenotypes in skeletal muscle and the central nervous system have been described in another FKRP mutant KI mouse (FKRP-NeoY307N) [79]. Abnormal brain development was shown in GTDC2-KO, POMTGnT1-KO, and brain-selective POMT2-cKO mice [71–73, 80]. These models are genetically engineered mice, but there is a spontaneous Large mutant mouse (Large myd) that exhibits abnormal α-DG glycosylation and shows skeletal muscle pathology and neuronal migration defects that recapitulate α-DGpathy phenotypes [38, 81]. This is probably the most used model of α-DGpathy.
Table 2.2
Summary of dystroglycanopathy mouse models introduced in this chapter
Model mice | Gene | Phenotypes | References |
|---|---|---|---|
Knockout (KO) | |||
POMT1 KO | POMT1 | Embryonic lethal | [69] |
Fukutin KO | FKTN | Embryonic lethal | [70] |
GTDC2 KO | GTDC2 | Postnatal lethality Brain abnormality | [71] |
POMGnT1 KO | POMGNT1 | Variable life span Brain abnormality | |
Chimeric mice | |||
Fukutin chimera | FKTN | Severe muscular dystrophy Brain and eye abnormality | [74] |
Knock-in (KI) | |||
Fukutin KI (retrotransposon) | FKTN | No obvious phenotype | [75] |
FKRP E310del homozygous | FKRP | Embryonic lethal | [78] |
FKRP P448L homozygous | FKRP | Muscular dystrophy Brain and eye abnormality | [78] |
FKRP-NeoTyr307Asn homozygous | FKRP | Postnatal lethality Reduced muscle mass Brain and eye abnormality | [79] |
Conditional KO (cKO) | |||
Fukutin-cKO (muscle precursor cell) | FKTN | Severe muscular dystrophy | |
Fukutin-cKO (myofiber) | FKTN | Mild muscular dystrophy | |
POMT2-cKO (brain) | POMT2 | Brain abnormality | [80] |
Spontaneous mutant
Related posts: Toward Regenerative Medicine for Muscular Dystrophies
Therapeutic Approach of iPS Cell Technology for Treating Muscular Dystrophy Toward Regenerative Medicine for Muscular Dystrophies
Therapeutic Approach of iPS Cell Technology for Treating Muscular Dystrophy
 Molecular Pathogenesis and Therapeutic Strategy in GNE Myopathy Molecular Pathogenesis and Therapeutic Strategy in GNE Myopathy
 Stem Cell-Based Therapy for Duchenne Muscular Dystrophy Stem Cell-Based Therapy for Duchenne Muscular Dystrophy
 Translational Research on DMD in Japan Translational Research on DMD in Japan
 Patient Registries for International Harmonized Clinical Development Patient Registries for International Harmonized Clinical Development
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|