CHAPTER 4 Wound-healing physiology
1. Compare and contrast wound-healing processes for each type of closure: primary intention, secondary intention, and tertiary intention.
2. Distinguish between partial-thickness wound repair and full-thickness wound repair, addressing the key components, phases, usual time frames, and wound appearance.
3. Explain the difference between acute and chronic full-thickness wounds.
4. Describe four characteristics of a chronic wound.
5. Describe the role of the following cells in the wound repair process: platelets, polymorphonuclear leukocytes, macrophages, fibroblasts, endothelial cells, and keratinocytes.
6. Explain how bioactive agents (e.g., growth factors and cytokines) and extracellular matrix proteins (e.g., matrix metalloproteinases and tissue inhibitors of metalloproteinases) help to regulate the repair process.
7. Describe features and characteristics of scarless healing.
8. Distinguish between keloid and hypertrophic scarring and the treatment of each.
9. Identify conditions and comorbidities that affect the wound-healing process.
For centuries, wound healing was regarded as a mysterious process, with wound management based on practitioner preference as opposed to scientific principles. Research over the past 3 decades has contributed much information regarding the wound-healing process and the factors that facilitate this process. We now know that repair is an extremely complex process involving hundreds, possibly thousands, of overlapping and “linked” processes (Goldman, 2004). It is critical for all wound care clinicians to base their interventions and recommendations on current data and to remain abreast of new findings and their implications for care. This chapter reviews the process of wound healing and discusses the implications for wound management.
Mechanism of wound healing
The ability to repair tissue damage is an important survival tool for any living organism. Regardless of the type or severity of injury, repair occurs by only two mechanisms—regeneration, or replacement of the damaged or lost tissue with more of the same, and scar formation, replacement of damaged or lost tissue by connective tissue that lacks some of the functions of the original tissue. Regeneration is the preferred mechanism of repair because normal function and appearance are maintained. Scar formation is a less satisfactory alternative and occurs only when the tissues involved are incapable of regeneration. Many invertebrate and amphibian species have the ability to regenerate entire limbs. Humans have only limited capacity for regeneration, and most wounds heal by scar formation (Calvin, 1998; Mast and Schultz, 1996; Wilgus, 2007).
In humans, the mechanism of repair for any specific wound is determined by the tissue layers involved and their capacity for regeneration. Wounds that are confined to the epidermal and superficial dermal layers heal by regeneration because epithelial, endothelial, and connective tissue can be reproduced. In contrast, deep dermal structures (e.g., hair follicles, sebaceous glands, sweat glands), subcutaneous tissue, muscle, tendons, ligaments, and bone lack the capacity to regenerate; therefore loss of these structures is permanent, and wounds involving these structures must heal by scar formation (Martin, 1997; Mast and Schultz, 1996).
The standard repair mechanism for human soft tissue wounds is connective tissue (scar) formation; however, an exception to this rule is the early-gestation fetus. Intrauterine surgical procedures performed during the second trimester result in scarless healing, a phenomenon that has been consistently reproduced in laboratory studies. Interestingly, the fetus loses the ability to heal without scarring at approximately 22 to 24 weeks’ gestation. Repair during the third trimester and the postnatal period follows the “usual” rules for repair and results in scar formation. A number of differences in the molecular environment for repair in the early-gestation fetus are thought to contribute to scarless repair and are discussed throughout this chapter (Bullard et al, 2002; Dang et al, 2003; Wilgus, 2007; Yang et al, 2003).
Partial thickness/full thickness
Partial-thickness wounds involve only partial loss of the skin layers, that is, they are confined to the epidermal and superficial dermal layers. Full-thickness wounds involve total loss of the skin layers (epidermis and dermis) and frequently involve loss of the deeper tissue layers as well (subcutaneous tissue, muscle, and bone) (see Figure 3-1). The time frame for repair and the repair process itself differ significantly for partial-thickness and full-thickness wounds.
Acute/chronic
Acute wounds typically are traumatic or surgical in origin. Acute wounds occur suddenly, move rapidly and predictably through the repair process, and result in durable closure. In contrast, chronic wounds fail to proceed normally through the repair process. Chronic wounds frequently are caused by vascular compromise, chronic inflammation, or repetitive insults to the tissue, and they either fail to close in a timely manner or fail to result in durable closure (Brissett and Hom, 2003; Clark, 2002; Pradhan et al, 2009).
Primary, secondary, tertiary
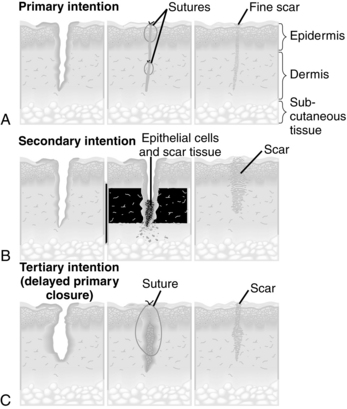
Classification of repair as primary-, secondary-, or tertiary-intention healing is based on the ideal of primary surgical closure for all wounds. Primary closure minimizes the volume of connective tissue deposition required for wound repair and restores the epithelial barrier to infection (Figure 4-1, A). Approximated surgical incisions are said to heal by primary intention; they usually heal quickly, with minimal scar formation, as long as infection and secondary breakdown are prevented. Wounds that are left open and allowed to heal by scar formation are classified as healing by secondary intention (Figure 4-1, B). These wounds heal more slowly because of the volume of connective tissue required to fill the defect. They are more subject to infection because they lack the epidermal barrier to microorganisms until late in the repair process. These wounds are characterized by prolongation of the inflammatory and proliferative phases of healing. Chronic wounds such as pressure ulcers and dehisced incisions typically heal by secondary intention. Wounds managed with delayed closure are classified as healing by tertiary intention, or delayed primary closure (Figure 4-1, C). This approach is sometimes required for abdominal incisions complicated by significant infection (Gabriel et al, 2010). Closure and/or approximation of the wound is delayed until the risk of infection is resolved and the wound is free of debris.
Wound-healing process
Wound healing is best understood as a cascade of events. Injury sets into motion a series of physiologic responses that are coordinated and sequenced and that, under normal circumstances, result in durable repair. Critical factors in the repair process include the cells that establish a clean wound bed and generate the tissue to fill the defect, the bioactive molecules that control cellular activity (growth factors and cytokines), and the wound-healing environment (extracellular matrix [ECM]). The wound-healing environment (matrix) provides a scaffold that promotes cell migration, and it contains a number of substances that regulate the activity of growth factors and cytokines and promote cell migration (e.g., matrix metalloproteinases) (Cheresh and Stupack, 2008; Gill and Parks, 2008; Macri and Clark, 2009).
Partial-thickness wound repair
Partial-thickness wounds are shallow wounds involving epidermal loss and possibly partial loss of the dermal layer (Plate 1). Partial-thickness wounds typically are approximately 0.2 cm in depth. These wounds are moist and painful because of the loss of the epidermal covering and the resultant exposure of nerve endings. When the wound involves loss of the epidermis with exposure of the basement membrane, the wound base appears bright pink or red. In the presence of partial dermal loss, the wound base usually appears pale pink with distinct red “islets.” These “islets” represent the basement membrane of the epidermis, which projects deep into the dermis to line the epidermal appendages. These islands of epidermal basement membrane are important in partial-thickness wound healing because all epidermal cells are capable of regeneration, and each islet will serve as a source of new epithelium (Clark, 2002; Winter, 1979).
The major components of partial-thickness repair include an initial inflammatory response to injury, epithelial proliferation and migration (resurfacing), and reestablishment and differentiation of the epidermal layers to restore the barrier function of the skin (Monaco and Lawrence, 2003; Staiano-Coico et al, 2000; Winter, 1979). If the wound involves dermal loss, connective tissue repair (granulation tissue formation) will proceed concurrently with epithelial repair (see Table 4-1) (Jahoda and Reynolds, 2001).
TABLE 4-1 Partial-Thickness Repair
| Level of Injury (Tissue Layers Involved) | Primary Phases of Repair | Outcomes |
|---|---|---|
| Epidermal loss | Inflammation (brief) Epithelial resurfacing Restoration of normal epithelial thickness/repigmentation | No loss of function No scar No change in skin appearance or color |
| Possible partial dermal loss | Dermal repair (if dermal loss involved) | Restoration of rete ridges/dermal papillae when dermis involved |
Epidermal repair.
Tissue trauma triggers the processes that result in epidermal repair: an acute inflammatory response followed by epidermal mitosis and migration (Harrison, et al, 2006). The inflammatory response produces erythema, edema, and a serous exudate containing leukocytes. When this exudate is allowed to dry on the wound surface, a dry crust, commonly referred to as a “scab,” is formed. In partial-thickness wounds the inflammatory response is limited, typically subsiding in less than 24 hours (Winter, 1979).
Epidermal resurfacing begins as the inflammatory phase subsides and is dependent on two processes: proliferation of epidermal cells throughout the wound bed and lateral migration of the epidermal cells at the leading edge. In order to migrate laterally, the keratinocytes at the wound edge must undergo several changes. First they must acquire a migratory phenotype. This involves breakdown of their attachments to adjacent cells, the basement membrane, and the underlying dermis. These attachments normally prevent lateral migration and provide epithelial stability. The keratinocytes then must undergo cytoskeletal alterations that support lateral movement. These alterations include flattening of the cells at the advancing edge of epithelium and formation of protrusions known as lamellipodia, which attach to binding sites in the wound bed. The migratory keratinocytes then move across the wound bed by alternately attaching to the ECM and then detaching and reattaching at a more distal point (Chen and Abatangelo, 1999; Frank et al, 2002; Harrison et al, 2006; Henry and Garner, 2003; Monaco and Lawrence, 2003; Staiano-Coico et al, 2000; Werner and Grose, 2003).
The epithelial cells within the wound bed continue this pattern of lateral migration until they contact epithelial cells migrating from the opposite direction. Once the epithelial cells meet, lateral migration ceases; a phenomenon known as contact inhibition (Monaco and Lawrence, 2003; Staiano-Coico et al, 2000).
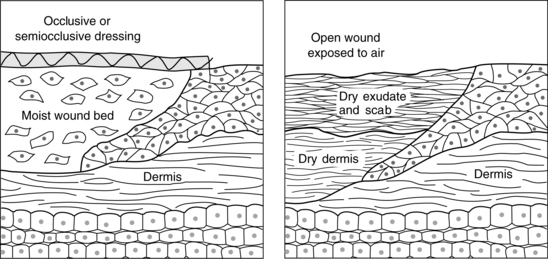
Epithelial resurfacing is supported by increased production of basal cells just behind the advancing edge and throughout the wound bed. The process typically peaks between 24 and 72 hours after injury. The rate of reepithelialization is affected by a number of factors of clinical significance. For example, resurfacing is promoted by maintenance of a moist wound surface. Winter (1979) found that partial-thickness wounds left open to air required 6 to 7 days to resurface, whereas moist wounds reepithelialized in 4 days. This is because cells can migrate much more rapidly in a moist environment (Figure 4-2). In contrast, when the surface of the wound is covered with a scab, migration is delayed while the epithelial cells secrete enzymes known as matrix metalloproteinases (MMPs). The MMPs lift the scab by cleaving the bonds that attach the scab to the wound bed, creating a moist pathway that supports keratinocyte migration (Clark, 2002; Harrison et al, 2006; Monaco and Lawrence, 2003; Staiano-Coico et al, 2000; Winter, 1979). Interestingly, hypoxic conditions in the wound bed also serve to stimulate keratinocyte migration, probably through increased production of the MMPs that promote lateral migration. This positive response to wound bed hypoxia is lost in the elderly, which may be one factor contributing to slower rates of reepithelialization in the aged (O’Toole et al, 2008). In contrast, high bacteria levels may serve as an impediment to reepithelialization. Data indicate that keratinocyte migration is inhibited by bacterial byproducts such as lipopolysaccharide (Loryman and Mansbridge, 2008). In vivo evidence strongly suggests that elevated glucose levels are another major impediment to epidermal proliferation and migration. This may be one factor contributing to delayed healing of superficial wounds in diabetic patients with poorly controlled glucose levels (Lan et al, 2008).
The newly resurfaced epithelium appears pale, pink, and dry in people of all races (see Plate 1). Because it is only a few cell layers thick, the new epithelium is very fragile and requires protection against mechanical forces such as superficial shear and friction.
Once epithelial resurfacing is complete, the epithelial cells resume vertical migration and epidermal differentiation so that normal epidermal thickness and function are restored. The normal anchors to adjacent epidermal cells and to the basement membrane are reestablished (Harrison et al, 2006). The “new” epidermis gradually repigments, matching the individual’s normal skin tone (see Plate 1). This is clinically relevant, as the wound is not completely healed until repigmentation has occurred (Monaco and Lawrence, 2003).
Regulatory factors.
The processes of epithelial proliferation and migration are regulated by a complex interplay between various MMPs and selected growth factors. Specifically, lateral migration is dependent on normal levels and function of multiple MMPs, which serve to break the attachments that bind the edge keratinocytes to adjacent cells and the underlying wound bed. In addition to playing a key role in establishment of a migratory phenotype, MMPs facilitate continued resurfacing by assisting the migrating keratinocytes to detach from the wound bed. Repetitive detachment and distal reattachment are critical to the resurfacing process (Gill and Parks, 2008; Lan et al, 2008). Epithelial proliferation is dependent in part on keratinocyte attachment to the ECM and in part on exposure to growth factors. Keratinocyte attachment is required for the cell to exit from the G0 phase of mitosis, and growth factors are required to stimulate cellular reproduction. A number of growth factors promote keratinocyte proliferation. They include transforming growth factor-α (TGF-α), keratinocyte growth factor, platelet-derived growth factor (PDGF), and epidermal growth factor (EGF). PDGF and EGF seem to be of particular importance. PDGF promotes keratinocyte migration early postinjury, and EGF seems to play an important role throughout the epithelialization process (Braiman-Wiksman et al, 2007; Myers et al, 2007; Schneider et al, 2008). Insulin-like growth factor may also play a role. An animal study demonstrated that exogenous application of insulin to partial-thickness wounds produced a significant increase in both epithelial proliferation and migration rate (Liu et al, 2009). See Chapter 20 for further discussion of the molecular factors regulating wound repair.
Dermal repair.
In wounds involving both dermal and epidermal loss, dermal repair proceeds concurrently with reepithelialization. By the fifth day after injury, a layer of fluid separates the epidermis from the dermal tissue. New blood vessels begin to sprout, and fibroblasts become plentiful by about the seventh day. Collagen fibers are visible in the wound bed by the ninth day. Collagen synthesis continues to produce new connective tissue until about 10 to 15 days after injury. This new connective tissue grows upward into the fluid layer. At the same time, the flat epidermis falls down around the new vessels and collagen fibers, recreating ridges at the dermal–epidermal junction. As the new connective tissue gradually contracts, the epidermis is drawn close to the dermis (Winter, 1979). Insulin and insulin-like growth factor may contribute to reformation of the dermal–epidermal junction. An animal study by Liu et al (2009) demonstrated more epidermal rete ridges and dermal papillae in wounds treated with exogenous insulin.
Full-thickness wound repair: wounds healing by primary intention
Full-thickness wounds, by definition, extend at least to the subcutaneous tissue layer and possibly as deep as the fascia–muscle layer or bone (see Plate 2). Full-thickness wounds may be either acute or chronic. This section addresses acute wound healing by primary intention, such as a surgical incision.
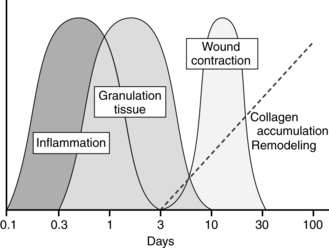
Many steps are involved in full-thickness repair, but they are commonly conceptualized as four major phases: hemostasis, inflammation, proliferation, and remodeling. Considerable overlap exists among the phases, and the cells involved in one phase produce the chemical stimuli and substances that serve to move the wound into the next phase. Thus normal repair is a complex and well-orchestrated series of events (Figure 4-3) and is affected by a number of systemic and local factors, including the wound environment/ECM.
Hemostasis.
Any acute injury extending beyond the epidermis causes bleeding, which activates a series of overlapping events designed to control blood loss, establish bacterial control, and seal the defect. Immediately upon injury, disruption of blood vessels exposes the subendothelial collagen to platelets, which trigger platelet activation and aggregation. Simultaneously, injured cells in the wound area release clotting factors that activate both the intrinsic and extrinsic coagulation pathways. As part of the coagulation pathway, circulating prothrombin is converted to thrombin, which is used to convert fibrinogen to fibrin. The end result is formation of a clot composed of fibrin, aggregated platelets, and blood cells. Hemostasis is further accomplished by a brief period of vasoconstriction mediated by thromboxane A2 and prostaglandin 2-α, substances released by the damaged cells and activated platelets. Clot formation seals the disrupted vessels so that blood loss is controlled. The clot also provides a temporary bacterial barrier, a reservoir of growth factors, and an interim matrix that serves as scaffolding for migrating cells (Brissett and Hom, 2003; Monaco and Lawrence, 2003; Phillips, 2000; Pullar et al, 2008; Werner and Grose, 2003).
Clot formation, followed by fibrinolysis (clot breakdown), is a critical event in the sequence of wound healing (Figure 4-4). The activation and degranulation of platelets cause the α-granules and dense bodies of the platelets to rupture, releasing a potent “cocktail” of energy-producing compounds and cytokines/growth factors (including complement factor C5a). These substances attract the cells needed to begin the repair process. They also provide fuel for the energy-intensive process of wound healing. The platelet-derived substances thought to be most critical to repair include PDGF, TGF-β, and fibroblast growth factor-2. Thus hemostasis, which is the body’s normal response to tissue injury, actually initiates the entire wound-healing cascade. The importance of hemostasis to wound healing is underscored by the finding that inadequate clot formation is associated with impaired wound healing and that the extrinsic coagulation pathway is critical to repair (Brissett and Hom, 2003; Frank et al, 2002; Monaco and Lawrence, 2003; Pullar et al, 2008; Samuels and Tan, 1999; Staiano-Coico et al, 2000).
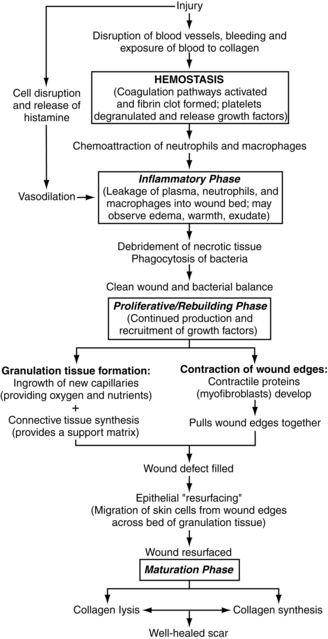
Inflammation.
Once the bleeding is controlled, the focus becomes control of infection and establishment of a clean wound bed. This process can be compared to the repair of a damaged home or building. Before rebuilding can begin, the damaged components must be removed. Wound “cleanup” involves breakdown of any devitalized tissue and elimination of excess bacteria by a team of white blood cells. Acute wounds, such as sutured lacerations or incisions, typically have limited devitalized tissue and low bacterial loads, so the cleanup phase usually is brief, lasting only hours to a few days (Braiman-Wiksman et al, 2007; Pradhan et al, 2009).
Within 10 to 15 minutes following injury, vasoconstriction subsides, followed by vasodilation and increased capillary permeability. Vasoactive substances released by damaged cells and by clot breakdown (histamine, prostaglandins, complement factor, thrombin) mediate this response. The dilated capillaries permit plasma and blood cells to leak into the wound bed. Clinically, this process is observed as edema, erythema, and exudate. At the same time, the damaged cells and platelets from the injured vessels produce cytokines and growth factors that attract leukocytes (neutrophils, macrophages, lymphocytes) to the wound bed. The twin processes of chemoattraction and vasodilation result in the delivery of multiple phagocytic cells to the wound site within minutes of injury (Gill and Parks, 2008; Rodriguez et al, 2008; Pradhan et al, 2009).
Leukocyte migration out of the vessels and into the wound bed occurs via margination and diapedesis. Margination involves adherence of the leukocytes to the endothelial cells lining the capillaries in the wound bed. Integrins on the surface of the leukocytes attach to cell adhesion molecules on the surface of the endothelial cells, causing the leukocytes to “line up” against the vessel wall. Through a process known as diapedesis, the leukocytes migrate through the dilated capillaries into the wound bed (Cross and Mustoe, 2003; Monaco and Lawrence, 2003; Wilgus, 2007).
The first leukocytes to arrive in the wound space, the neutrophils, are present in the wound bed within minutes following injury and dominate the scene for the first 2 to 3 days. The primary function of neutrophils is phagocytosis of bacteria and foreign debris. Neutrophils first bind to the damaged tissue or target bacteria via cell adhesion molecules and then engulf and destroy the target molecules via intracellular enzymes and free oxygen radicals. In addition, growth factors released by the neutrophils attract additional leukocytes to the area (Cross and Mustoe, 2003; Goldman, 2004; Monaco and Lawrence, 2003; Pullar et al, 2008; Schultz et al, 2003).
By days 3 to 4 after injury, the neutrophils begin to spontaneously disappear as a result of apoptosis and are replaced by macrophages (activated monocytes). The macrophages continue to phagocytize bacteria and break down damaged tissues as already described. In addition, the macrophages release a large number of potent growth factors that stimulate angiogenesis, fibroblast migration and proliferation, and connective tissue synthesis (Pullar et al, 2008; Rodriguez et al, 2008).
T lymphocytes are present in the wound tissue in peak quantities between days 5 and 7 after injury. T lymphocytes contribute to the inflammatory phase of wound healing by secreting additional wound-healing cytokines and by destroying viral organisms and foreign cells. The elimination of these cells can delay or compromise the repair process (Monaco and Lawrence, 2003).
Although all leukocytes contribute to elimination of bacteria and establishment of a clean wound bed, macrophages contribute the most significantly to the repair process. Studies indicate that wounds can heal without neutrophils, especially if no bacterial contamination is present. However, elimination of macrophages severely compromises wound repair (Cross and Mustoe, 2003; Gabriel et al, 2010).
The result of the inflammatory phase of wound healing is a clean wound bed. In acute wounds healing by primary intention, the inflammatory phase lasts approximately 3 days. At this point, bacterial levels usually are controlled, and any devitalized tissue has been removed. Elimination of these noxious stimuli allows the wound to transition to the “rebuilding” phase. During this transition, the cells in the wound bed begin to produce growth factors that stimulate proliferation rather than inflammation (Goldman, 2004; Staiano-Coico et al, 2000). However, in wounds complicated by necrosis and/or infection, the inflammatory phase is prolonged and wound healing is delayed (Braiman-Wiksman et al, 2007; Clark, 2002). A prolonged inflammatory phase increases the risk for wound dehiscence because approximation of the incision is totally dependent upon the closure material (sutures, staples, fibrin glue) until sufficient connective tissue has been synthesized to provide tensile strength to the incision. (Tensile strength during the inflammatory phase is 0%.)
Factors affecting the duration and intensity of inflammation.
The intensity and duration of the inflammatory phase appear to be critical factors in the amount of scar tissue produced. Numerous studies support a strong link between prolonged inflammation and hyperproliferative scarring (Dubay and Franz, 2003; Pradhan et al, 2009; Pullar et al, 2008; Rahban and Garner, 2003; Robson, 2003). Thus the clinician needs to have a clear understanding of both proinflammatory and antiinflammatory factors and the implications for wound management. As noted, local factors such as bacterial loads and presence of devitalized tissue are major proinflammatory factors that should be aggressively managed through debridement and appropriate use of antimicrobial agents. The ECM is another important determinant and can either promote or inhibit inflammation, depending on the types of MMPs produced and their functions. Some MMPs promote inflammation by releasing proinflammatory cytokines from the cells in the wound bed; others inhibit inflammation by degrading proinflammatory cytokines or inhibiting their release (proinflammatory cytokines promote inflammation by continuing to attract inflammatory cells to the wound bed) (Pradhan et al, 2009). For example, tumor necrosis factor-α (TNF-α) is a proinflammatory cytokine that normally is present at high levels during the early inflammatory phase, and levels of TNF-α are controlled by two ECM proteins. One protein, a disintegrin and metalloproteinase (ADAM-17), promotes release of TNF-α. Another protein (tissue inhibitor of metalloproteinase-3 [TIMP-3]) controls ADAM-17, thereby reducing production of TNF-α. Research is ongoing into factors that determine the combination and concentration of ECM proteins and into strategies for measuring and controlling levels of various MMPs to achieve therapeutic outcomes (Gill and Parks, 2008; Macri and Clark, 2009).
Diabetes is a clinical condition associated with prolonged inflammation. Some data suggest that leukocyte migration may be impaired; in addition, the leukocytes that do migrate to the wound bed frequently are dysfunctional, especially in the presence of hyperglycemia. The end result of diminished leukocyte migration and impaired leukocyte function is failure to effectively control bacterial loads. This situation results in a persistent stimulus for proinflammatory cytokines, chronic inflammation, and delayed healing (Pradhan et al, 2009). Conditions resulting in ischemia/hypoxia also produce prolonged inflammation, because oxygen and reactive oxygen species (ROS) are required for oxidative killing of bacteria. The inability to control bacteria that accompanies hypoxia results in chronic inflammation and failure to heal. Hyperbaric oxygen therapy can improve tissue oxygen levels, contribute to bacterial control, and reduce the production of proinflammatory cytokines, thus promoting the repair process (Rodriguez et al, 2008; Thom, 2009).
Conditions and medications that affect the function of autonomic and sensory nerves and their receptors may also affect inflammation. For example, diabetes is associated with diminished production of substance P, a neuropeptide that normally contributes to a healthy inflammatory response by supporting vasodilation and leukocyte migration. Diminished production of substance P may be one factor leading to compromised inflammation and impaired healing in the diabetic population (Pradhan et al, 2009). From a therapeutic perspective, β-adrenergic agents may reduce inflammation by reducing neutrophil recruitment and production of inflammatory cytokines; further research on this topic is needed (Pullar et al, 2008).
Proliferation.
Epithelialization.
Epithelialization of a full-thickness wound healing by primary intention begins within hours after injury and typically is complete within 24 to 48 hours. This “neoepithelium” is only a few cells thick but is sufficient to provide a closed wound surface and a bacterial barrier. This process is the basis for the Centers for Disease Control recommendation that new surgical incisions be covered with a sterile dressing for the first 24 to 48 hours postoperatively. Until reepithelialization is complete, the potential for bacterial invasion exists, and the wound should be managed with sterile wound care and a cover dressing that provides a bacterial barrier (Cross and Mustoe, 2003; Mangram et al, 1999; Monaco and Lawrence, 2003). As with partial-thickness wound repair, the processes of lateral migration, vertical migration, and differentiation continue throughout the proliferative phase and gradually reestablish epidermal thickness and function (Braiman-Wiksman et al, 2007; Myers et al, 2007). In full-thickness wounds, the new epidermis is slightly thinner than the original epidermis. Because the neoepidermis rather than normal dermis is covering scar tissue, the rete pegs that normally dip into the dermis are lacking (Monaco and Lawrence, 2003).






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


