Fig. 2.1
Histology of early OA (a–c) and RA (d–f) samples, indicating thickened synovium and infiltration of inflammatory cells. The histology changes in early OA can be quite suggestive of inflammation, even resembling the changes seen in RA
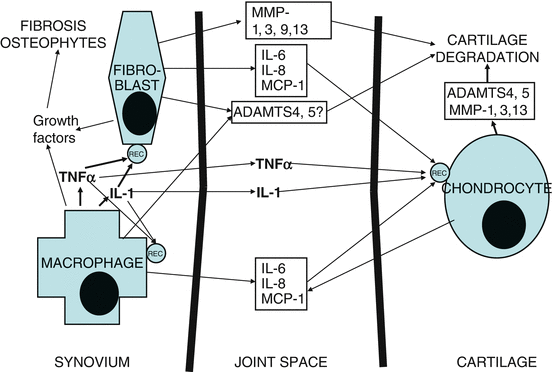
Fig. 2.2
A simplified view of cell signalling in the OA synovium and the production of inflammatory and destructive mediators
Rheumatoid arthritis (RA) is the most common of the inflammatory arthritides and a both important and treatable disease. It is characterised by intense synovial inflammation and proliferation, giving rise to cartilage destruction and bony erosions. In RA, it is today accepted that both inflammatory and destructive features of the disease are driven through synovitis. The RA synovium has a plentiful infiltrate of activated macrophages, particularly at the cartilage-pannus junction [1]. These macrophages produce tumour necrosis factor-α (TNFα), interleukin (IL)-1β and other proinflammatory cytokines. Since there is a ‘cytokine cascade’ with TNFα driving many of the other inflammatory mediators, this cytokine has become a key therapeutic target in RA, with several anti-TNFα biologic agents being used with considerable success [2]. Although biologics with anti-B-cell and anti-T-cell co-stimulation properties have since been introduced, as well as anti-IL6-receptor antibodies, the anti-TNFα agents remain a mainstay of RA therapy, with long-term sustained efficacy and safety.
Osteoarthritis (OA), one of the most common diseases among mammals, is characterised pathologically by focal areas of damage on articular cartilage centred on load-bearing areas, associated with formation of new bone at the joint margins and changes in subchondral bone. Given the huge economic and personal burden of OA, and the fact that this disease is the major cause for the increasing demand for joint replacements, there is urgent need for disease-modifying treatments to stop or at least slow the development and progression of OA. Various drug candidates have been brought forward, but hitherto, none of them has been clearly demonstrated to have clinical benefit in OA [3, 4]. Nor have the widely available ‘nutraceuticals’ glucosamine and chondroitin sulphate been demonstrated to have benefit in this disease [5].
The role of macrophages driving inflammatory and destructive pathways in RA is still controversial. Clinically, OA patients have a variable degree of synovitis, sometimes with quite aggressive large-joint arthritis with exudation. There is evidence that this synovitis is macrophage driven and that it contributes to disease progression in early stages of OA. When OA is clinically significant, disease is already well advanced, however, and clinical trials of strategies directed against macrophage-produced cytokines have not been successful. The concept of inflammatory synovitis contributing to OA pathology has attracted interest for some time [6–8], particularly the role of activated synovial macrophages in driving inflammatory and destructive pathways in the OA synovitis [9, 10], but the path from the introduction of this concept to its utilisation for the potential development of a disease-modifying anti-osteoarthritic drug remains a lengthy one.
This chapter will give an overview of some important recent findings concerning the ability of macrophages to drive inflammatory and destructive disease mechanisms in RA and OA, the role of their proinflammatory cytokines in doing so and the potential for macrophages and macrophage-produced cytokines to be used as therapeutic targets for the development of disease-modifying drugs. The role of synovial macrophages in the mechanism of action of radiosynovectomy in RA and OA will also be discussed.
2.2 The Role of Macrophages in RA
RA is a common (prevalence 1 % in Europe and the USA) and potentially debilitating inflammatory disease, recognised in Europe since the early 1800s. It mainly involves joint inflammation but also has systemic manifestations in many patients, like rheumatoid nodules, anaemia and bone marrow changes, lung fibrosis, pleuritis and pericarditis, eye disease with scleritis or episcleritis and occasionally small vessel vasculitis. Clinically, RA presents with symmetrical inflammatory arthritis involving both large and small joints, typically the wrists, metacarpophalangeal joints and proximal interphalangeal joints, as well as early symmetrical involvement of the metatarsophalangeal joints. Typically, there is visible and palpable joint swelling, caused by synovial inflammation and hypertrophy, and exudation of excess of synovial fluid. In severe early RA, there is pronounced morning stiffness, sometimes lasting for hours, and generalised weakness, malaise and low-grade fever. If RA is left untreated, there will be progressive development of joint erosions involving both cartilage and subchondral bone, as well as joint contractures leading to permanent disability. These joint erosions can be seen on plain X-rays, and early on an MRI scan, and they thus have diagnostic value. Since erosions are irreversible, causing permanent structural damage, there is a need for a swift diagnosis for the disease to be challenged with the most potent disease-modifying antirheumatic drugs (DMARDs).
The American College of Rheumatology (ACR) has established a set of seven criteria (symmetrical arthritis, hand arthritis, polyarthritis, significant morning stiffness, erosions, rheumatoid nodules and rheumatoid factor positivity) that are both useful and generally accepted. As soon as these criteria are fulfilled, the practice is to institute treatment with one of the most potent DMARDs: methotrexate, sulphasalazine or leflunomide. Therapeutic concentrations of some of the old-fashioned DMARDs, like auranofin and the antimalarials chloroquine and hydroxychloroquine, appear to have an effect on macrophage-produced proinflammatory cytokines [11], although nontoxic concentrations of methotrexate or leflunomide do not [11, 12]. The problems with these DMARDs are that they are not sufficiently potent in many patients, that they have severe and frequent side effects and that it is impossible to predict the response to a certain DMARD. Partial control of inflammation in RA does not appear to prevent joint damage in RA, and the majority of patients with active disease become disabled within 20 years. For those with active disease or extra-articular manifestations, the mortality is comparable to that of patients with three-vessel coronary arteriosclerosis or stage IV Hodgkin’s disease [13, 14].
The inflamed RA synovium contains a mixed population of cells. Apart from the synovial fibroblasts, there are activated B and T cells, plasma cells, mast cells and activated macrophages. These cells have all been recruited via a neovascularisation process with associated lymphangiogenesis. In RA, the synovial lining, normally just two to three cells thick, becomes eight to ten cells thick, with infiltration of synovial fibroblasts and macrophages. The sublining area of the synovium, which normally has relatively few cells, becomes heavily infiltrated with inflammatory cells, macrophages prominent among them. This accumulation of cellular infiltrate is accompanied by neoangiogenesis, with an extensive network of newly formed blood vessels. The hypertrophied pannus invades and destroys cartilage and subchondral bone, and activated macrophages are particularly numerous at the cartilage-pannus junction. A study of synovial biopsies showed a clear correlation between macrophage activation markers and the general inflammatory activity [15]. Another study observed a correlation between macrophage numbers in the lining and sublining layers of the rheumatoid synovium and the subsequent development of erosions [16]. Macrophages expressing the cytokines IL-1α and TNFα are prominent near the cartilage-pannus junction [17]. Apart from their antigen-presenting capacity, these macrophages probably do not have a direct causal pathogenic effect in RA, but they remain key players in mediating inflammation and joint destruction in acute and chronic RA, as mediators of proinflammatory, destructive and tissue remodelling in this disease.
The RA synovial macrophages are in a highly activated state, overexpressing a number of cytokines that mediate local and systemic inflammation and tissue remodelling [1]. They also overexpress chemokines like IL-8, macrophage inflammatory protein 1 and monocyte chemoattractant protein 1, which stimulate migration of inflammatory cells and stimulate angiogenesis. Although the synovial fibroblasts are the main producers of matrix metalloproteinases (MMPs) in RA, macrophages can produce MMP-9 and MMP-12, as well as tissue inhibitor of metalloproteases 1, which attempts to control excessive tissue destruction. In RA, it is today accepted that the synovitis is mainly cytokine driven, through a disequilibrium between proinflammatory (TNFα, IL-1) and anti-inflammatory (IL-10, the IL-1 receptor antagonist, soluble TNF receptors) cytokines. These proinflammatory cytokines are largely produced by synovial macrophages. A key early observation was when cocultures of rheumatoid synovial cells were treated with neutralising anti-TNF antibodies, the production of another key proinflammatory cytokine, IL-1, practically ceased [18]. Further experiments showed that other proinflammatory cytokines, like IL-6, IL-8 and GM-CSF, were also driven by TNF in RA synovial cell cocultures [19–21]. These observations laid the foundation for the concept of macrophage-produced TNFα as the main mediator of disease, driving the other proinflammatory cytokines through occupying a key position at the apex of a cytokine cascade.
The hypothesis of TNFα as the dominant proinflammatory mediator in rheumatoid inflammation was tested in a model of murine collagen-induced arthritis, using either specific anti-TNF monoclonal antibodies or a soluble TNF receptor fused to the immunoglobulin Fc fragment, both of which potently ameliorated clinical symptoms of arthritis and prevented joint destruction [22, 23]. The first clinical trial of anti-TNF therapy in RA was begun in 1992, using the chimeric anti-TNF monoclonal antibody infliximab. This short-term open-label trial enrolled 20 patients, and although it was primarily intended to test dosing and safety, patients experienced a marked reduction in pain, stiffness and joint swelling within 24 h [24]. A subsequent multicentre, placebo-controlled, randomised double-blind study in 73 patients with active RA used a single intravenous infusion of either placebo, 1 mg/kg of infliximab, or 10 mg/kg of infliximab. An intention-to-treat analysis showed that only 2 of 24 placebo patients responded after 4 weeks, versus 11 of 25 patients treated with 1 mg/kg infliximab and 19 of 24 patients treated with 10 mg/kg infliximab. In patients receiving the higher dose, response duration was 8 weeks. There were impressive reductions in tender joint count, as well as in laboratory markers of disease activity, such as IL-6, CRP and ESR [25]. In a longer-term multicentre placebo-controlled, randomised, double-blind phase II study in 101 RA patients who had active disease in spite of methotrexate treatment, patients who received 3 or 10 mg/kg infliximab again showed excellent responses, although patients who received 1 mg/kg had a shorter duration of response. There was a clear synergy between infliximab and methotrexate in this study, indicating different mechanisms of action for these two compounds [26]. These data provided the rationale for a randomised phase III study of infliximab, the Anti-TNF Therapy of Rheumatoid Arthritis with Concomitant Methotrexate (ATTRACT) trial, a multicentre study involving 428 patients with active RA in spite of methotrexate treatment. The patients were split into five groups: one receiving placebo, the other four either 3 or 10 mg/kg infliximab at four-weekly or eight-weekly intervals. Again, benefit from infliximab was both swift and pronounced, with 60–70 % improvement in the tender joint count and normalisation of the CRP after the first infusion [27]. The medication was well tolerated, and the response was sustained until the 54-week end point of the study. In this trial, it was also possible to evaluate the development of joint damage through modified Sharp scoring of radiographs of the hands and feet. In the 88 patients treated with methotrexate and placebo, the median increase in this index was 4.0, but in the 340 patients treated with methotrexate and different regimens of infliximab, the score was unchanged. Thus, control of RA inflammation by infliximab prevented cartilage degradation and bony erosions [28].
As a consequence of the pivotal clinical studies quoted above, neutralisation of TNFα, the most important proinflammatory cytokine in the rheumatoid synovium, has become the preferred strategy to modulate macrophage function in RA. Infliximab (Remicade) is today one of the established biologics for the treatment of RA. The second anti-TNF therapeutic strategy in RA is the p75 TNF receptor Ig fusion protein etanercept (Enbrel), for which clinical studies were completed in 1998, with benefit similar to that observed with infliximab [29, 30]. The third available anti-TNF biologic was adalimumab (Humira), a fully human anti-TNFα antibody; it was followed by the PEGylated certolizumab pegol (Cimzia) and by golimumab (Simponi). All these five biologics have been widely approved for use on both sides of the Atlantic and are commercially available; there are even more anti-TNF biologics being developed, one of them the PEGylated soluble TNF receptor pegsunercept, as well as ‘biosimilars’ or follow-on biologics, which are likely to have an impact in the near future. Long-term data has shown that these anti-TNF biologics are both safe and effective: in the majority of patients, response is sustained over a 5-year period, or longer [31–33]. If patients are chosen wisely, and frail and elderly people avoided, along with those at risk for opportunistic infections, and careful screening for latent tuberculosis performed prior to treatment initiation, the drugs are perfectly safe even for long-term use. Indications for the anti-TNF biologics have widened from RA and Crohn’s disease to encompass other forms of arthritis, such as severe or moderate psoriatic arthritis, early and severe ankylosing spondylitis and polyarticular juvenile idiopathic arthritis; some of them have plaque psoriasis and ulcerative colitis as indications, and they are undergoing clinical trials in a variety of inflammatory disorders. Regulatory authorities have been forced to ‘ration’ the anti-TNF biologics, and the current recommendation from the UK National Institute of Clinical Excellence is that to qualify for treatment with an anti-TNF biologic, patients must have failed two DMARDs, one of which must be methotrexate.
The development of biologic anti-TNF therapy against RA and other inflammatory diseases invigorated the search for other therapeutic targets in RA, with much money invested from the pharmaceutic industry, and some very promising results. The biologic rituximab (MabThera), an anti-CD20 antibody targeting B cells, which was previously used for B-cell lymphoma, has been proven to be safe and effective in RA. Several other anti-B-cell biologics, like ocrelizumab, ofatumumab and atacicept, are in clinical development. The humanised monoclonal antibody tocilizumab (RoActemra) that binds to membrane-bound and soluble forms of the IL-6 receptor has recently been found superior to adalimumab in monotherapy in a phase 4 trial in RA [34]. The anti-IL-6 antibody sarilumab and the anti-IL-6 receptor clazakizumab are both in clinical development. Another novel strategy concerns blocking T-cell co-stimulation via the anti-CTLA4 antibody abatacept (Orencia). There are many more antirheumatic drugs in development, both biologics and small molecules. The monoclonal antibodies secukinumab and brodalumab target the IL-17 family of cytokines. The small molecule tofacitinib (Xeljanz) is a Janus kinase (JAK) 3 inhibitor, which has been approved for the treatment of moderate to severe RA in the USA but not in the UK or in continental Europe. Baricitinib and ruxolitinib are two JAK 1/2 inhibitors in clinical development.
At the present time, making use of an anti-TNF biologic to block TNFα may well be the safest and most effective way to modulate macrophage function in RA, but much research has gone into investigating other options. For several decades, there has been a search for small-molecule inhibitors of TNFα production. Since it is known that in RA, the spontaneous production of TNFα from the synovial macrophages is NFκB dependent [35], there has been interest in inhibitors of this transcription factor, although NFκB is too ubiquitous a transcription factor for systemic inhibition to be clinically feasible. There has been interest in intra-articular administration of NFκB inhibitors, however, although this would only have a monoarticular effect [36, 37]. The phosphodiesterase 4 inhibitor apremilast (Otezla) has been reported to inhibit the spontaneous production of TNFα from RA synovial macrophages [38]. It is approved for the treatment of psoriatic arthritis in the USA but not as yet for RA. There is a good deal of recent literature on macrophages in RA, and the potential to induce macrophage apoptosis, to deplete synovial macrophages, or to change the macrophage polarisation from a M1 (classical, inflammatory) to M2 (alternative, anti-inflammatory) [39–41]. It has been demonstrated that RA synovial lining macrophages express folate receptor-β, and there has been an interest in targeting this to administer either folate antagonists [42] or an immunotoxin reducing the number of macrophages and inducing benefit in an animal model of arthritis [43].
2.3 The Role of Macrophages in OA
Clinically, RA and OA are usually easy to differentiate. In RA, X-rays of affected joints show erosions and periarticular osteoporosis, whereas in OA, they show reduction of joint space as a sign of cartilage degradation and, in later stages of the disease, bony sclerosis and osteophytes. The joint pattern differs, with early RA affecting the proximal interphalangeal, metacarpophalangeal and metatarsophalangeal joints and OA usually affecting the large joints, like the hips and knees, and also the distal interphalangeal joints. RA patients have an elevated erythrocyte sedimentation rate and C-reactive protein; the vast majority of OA patients do not. In RA patients, synovitis is a major feature of the disease, causing joint swelling and exudation and driving cartilage degradation and the formation of pannus and erosive changes. In OA, there is much less joint swelling and exudation and no pannus or erosions. But still, many OA patients have a variable degree of synovitis. Some of them may develop quite aggressive inflammatory OA of the knee or hip joint, sometimes with marked exudation, which can be helped by arthrocentesis and injection of local steroids. Synovial inflammation is likely to contribute to disease progression in OA, as judged by the correlation between biological markers of inflammation and the progression of structural changes in OA [44, 45]. Histologically, the OA synovium shows hyperplasia with an increased number of lining cells and a mixed inflammatory infiltrate mainly consisting of macrophages [46]. Synovial biopsies from patients with early inflammatory OA may even resemble RA biopsies morphologically, although the percentage of macrophages is lower (1–3 % as compared with 5–20 %) and the percentages of T and B cells much lower [47–49]. The marked differences in cell percentages in the inflammatory infiltrate between RA and OA would speak in favour of differences also in the cytokine interdependence in these two diseases. For example, the great scarcity of T cells in the OA synovium would tend to rule them (and their cytokines) out as potential drivers of synovitis in this disease. The synovial fluid of patients with active RA synovitis and effusion contains numerous polymorphonuclear leucocytes, something that is not the case in OA, another indicator that there is difference in pathophysiology between RA and OA synovitis. If it is accepted that synovial inflammation, and the production of proinflammatory and destructive mediators from the OA synovium, is of importance for the symptoms and progression of osteoarthritis, it is a key question which cell type in the OA synovium is responsible for maintaining synovial inflammation. In RA, macrophage-produced TNFα is a major therapeutic target, but much less is known about macrophage biology in OA, although histological studies have demonstrated that OA synovial macrophages exhibit an activated phenotype and that they produce both proinflammatory cytokines and vascular endothelial growth factor [46, 50].
The spontaneous production of a variety of pro- and anti-inflammatory cytokines, including TNFα, IL-1β and IL-10, is one of the characteristics of synovial cell cultures derived from digested RA or OA synovium. In addition, the major MMPs and TIMPs are spontaneously produced by these cell cultures [47, 48]. Less TNFα and IL-10 is produced from OA samples, but the levels are still easily detectable by ELISA [48]. It is possible to use effective adenoviral gene transfer in this model without causing apoptosis or disrupting intracellular signalling pathways. Using an adenovirus effectively transferring the inhibitory subunit IκBα, it was possible to selectively inhibit the transcription factor NFκB in synovial cocultures from RA or OA patients. Macrophage-produced TNFα and IL-1β were very strongly NFκB dependent in the RA synovium, but in OA synovium, adenoviral transfer of IκBα did not affect IL-1β production and had only a partial effect on TNFα. Effects on other cytokines were similar in RA and OA synovium, with IL-6 and IL-8 both being NFκB dependent, as well as the p75 soluble TNF receptor, whereas IL-10 and the IL-1 receptor antagonists were both NFκB independent. In addition, the matrix metalloproteinases (MMP) 1, 3 and 13 were strongly NFκB dependent in both RA and OA, but their main inhibitor, tissue inhibitor of metalloproteinases (TIMP)-1, was not [48]. The differential effect of NFκB downregulation on the spontaneous production of TNFα and IL-1β on RA and in OA would indicate that the regulation of at least one key intracellular pathway differs fundamentally between these diseases. It is known that both TNFα and IL-1β have functional NFκB elements on their promoters and that in various macrophage models, there are both NFκB-dependent and NFκB-independent ways of inducing TNFα and IL-1β [51, 52]. It would seem as if there are fundamental differences in the regulation of macrophage-produced TNFα and IL-1β between RA and OA, with cytokine levels being higher and NFκB playing a more important role in RA [48, 51, 53].
In the abovementioned model of cultures of osteoarthritis synovial cells, specific depletion of synovial macrophages could be achieved using incubation of the cells with anti-CD14-conjugated magnetic beads [54]. These CD14+-depleted cultures of synovial cells no longer produced significant amounts of macrophage-derived cytokines like TNFα and IL-1β. Interestingly, there was also significant inhibition (40–70 %) of several cytokines produced mainly by synovial fibroblasts, like IL-6 and IL-8, and also significant downregulation of MMP-1 and MMP-3. This would indicate that OA synovial macrophages play an important role in activating fibroblasts in these densely plated cultures of synovial cells and in perpetuating the production of proinflammatory cytokines and destructive enzymes. That the regulation is not tighter than observed is probably because the fibroblasts have an activated phenotype when put into culture, with considerable spontaneous production of cytokines and other mediators. It can be speculated that once the macrophages are removed, the synovial fibroblasts change their phenotype and downregulate their production of both proinflammatory cytokines and destructive MMPs. To investigate the mechanisms involved in this macrophage-driven stimulation of inflammatory and degradative pathways in the OA synovium, specific neutralisation of the endogenous production of TNFα and/or IL-1β was used in the cultures of OA synovial cell [54]. OA synovial cell cultures were either left untreated, incubated with the p75 TNF soluble receptor Ig fusion protein etanercept (Enbrel), incubated with a neutralising anti-IL-1β antibody or incubated with a combination of Enbrel and anti-IL-1β. As could be expected, TNFα production was effectively neutralised by Enbrel treatment and IL-1β by treatment with the neutralising anti-IL-1β antibody. There was no effect of Enbrel on IL-1β production nor did the neutralising anti-IL-1β antibody affect the production of TNFα. This is in marked contrast to the situation in RA, where IL-1β is strongly TNFα dependent in these cultures of synovial cells [18]. This finding would seem to indicate yet another difference in macrophage cytokine biology between RA and OA: whereas TNFα is the ‘boss cytokine’ in the RA synovium, regulating the production of IL-1β, there is a redundancy between these two cytokines in the OA synovium, with neither TNFα nor IL-1β regulating the production of the other.
Both Enbrel and the neutralising anti-IL-1β antibody inhibited IL-6 and IL-8, with 60 % inhibition achieved when both IL-1β and TNFα were neutralised. The production of MCP-1 was not affected by the neutralising anti-IL-1β antibody, but it was significantly decreased by Enbrel and by the combination of the two. It was also possible to study the effect of neutralising IL-1β and/or TNFα on the mRNA expression and protein production of the major MMPs and aggrecanases, using RT-PCR and ELISA analysis in parallel [54, 55]. The results indicate that although neither Enbrel nor the neutralising anti-IL-1β antibody had an impressive effect on the important collagenases MMP-1 and MMP-13, combination of the two led to significant inhibition both on the mRNA and protein levels. These findings indicate that in the OA synovium, the macrophages potently regulate the production of several important fibroblast-produced cytokines and MMPs, via a combined effect of IL-1β and TNFα. There was no effect of either Enbrel or the neutralising anti-IL-1β antibody on ADAMTS5 expression, nor was it at all affected by a combination of these treatments. Thus, ADAMTS5 appears to be constitutive in OA synovial cells. In contrast, ADAMTS4 was significantly (p < 0.05) inhibited by Enbrel and more potently (p < 0.01) inhibited by a combination of Enbrel and the neutralising anti-IL-1β antibody. This would indicate that in the human OA synovium, the upregulation of ADAMTS4 is dependent on TNFα and IL-1 produced by the synovial macrophages, whereas the level of ADAMTS5 is not changed by these cytokines [54, 55]. Thus, there is good evidence that in OA synovium and cartilage, ADAMTS4 is the aggrecanase induced by proinflammatory cytokines, whereas ADAMTS5 appears to be constitutive [54–58]. If it is accepted that OA is a cytokine-driven disease, as indicated by some recent papers suggesting that macrophage-produced IL-1 and TNF play a role in driving destructive responses in OA, this finding would render it likely that ADAMTS4 is the aggrecanase responsible for aggrecanolysis in OA. This is a finding of some importance for the debate regarding the major aggrecanase in OA, which is still ongoing. In murine models of degenerative joint disease, ADAMTS5 is the pathologically induced aggrecanase. Mice lacking ADAMTS4 develop normally and develop surgically induced OA in a similar manner to wild-type mice, but deletion of ADAMTS5 protects mice from developing OA [59–61]. However, there is a discrepancy between human and murine cells with regard to the regulation of ADAMTS4 [62, 63]. If the human, but not murine, ADAMTS4 gene responds to IL-1 stimulation, this brings into question the use of a murine model for the study of human aggrecanolysis, particularly since the normal function of at least ADAMTS5 appears to differ between rodents and primates, the enzyme having vital importance for versican turnover and myofibroblast differentiation in monkeys [64].
Related posts:
 Risk of Cancer Induction
Intra-articular Corticosteroid Treatment of Inflammatory Joint Diseases
Local Complications After Radiosynovectomy and Possible Treatment Strategies: A Literature Survey
Risk of Cancer Induction
Intra-articular Corticosteroid Treatment of Inflammatory Joint Diseases
Local Complications After Radiosynovectomy and Possible Treatment Strategies: A Literature Survey
 Synovectomy of Rheumatoid Joints
Synovectomy of Rheumatoid Joints
 Radiation Exposure of Medical Staff and Radiation Protection Measures
Radiation Exposure of Medical Staff and Radiation Protection Measures
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


