Fig. 11.1
Toluidine blue staining of a cartilage biopsy specimen overlying a bone bruise harvested from an ACL-injured patient before ACL reconstruction (from [6]). This specimen was histologically graded as an H-2 cartilage lesion (grading scale from H-0 (normal) to H-4 (moderate to severe fibrillation and complete loss to toluidine blue staining throughout the cartilage thickness)). Here, the articular surface is intact, but there is marked loss of matrix proteoglycans and noticeable cell death in the superficial zone
Macroscopically, four types of acute traumatic cartilage lesions were classified in a study of knee trauma [7] associated with shear or blunt compressive impact of cartilage. Tissue morphology ranged from stellate chondral fractures of cartilage to fissuring and milder fibrillation of the surface. Chondral lesions are common in the immature cartilage of children as well as in adolescents and adults and have been reported to be the most common injury to immature human knees [8].
In a recent study of ACL rupture patients 18–35 years old [5] using quantitative MRI imaging at the time of surgical reconstruction, T1ρ relaxation measurements indicated an acute increase in water content of the matrix after blunt trauma, consistent with tissue swelling. In another study of ACL-injured patients with MRI taken within 8 weeks of injury just prior to reconstruction, the cartilage overlying a bone bruise in the lateral tibia showed T1ρ signal changes occurring immediately after injury and at 1-year follow-up [9]. Longitudinal MRI assessment of the central weight-bearing aspects of medial and lateral femoral cartilage via dGEMRIC [10] suggested that GAG content was lower in ACL-injured knees than in a normal healthy reference cohort, both at 3 weeks and 2.3 years after injury. At the same time, measurement of cartilage and bone markers in the synovial fluid in the acute phase of ACL injury with hemarthrosis showed clear local biochemical responses to trauma, including increases in proinflammatory cytokines IL-1β, IL-6, IL-8, and TNFα [11]. Increases in the levels of several of these cytokines were also reported in previous reports [12–14].
The timeline of degradative events to cartilage after traumatic joint injury has been very well summarized by Lotz and Kraus [1] (see Table 11.1). After the immediate consequences of injury, progressive degeneration of cartilage is hypothesized to be stimulated by a combination of biomechanical factors (e.g., kinematic gait changes [15] that may occur with or without reconstructive surgery [16]) as well as cell biological and inflammatory mediators [11, 17]. These processes involve further changes in cartilage extracellular matrix and concomitant alterations in tissue biomechanical and physicochemical properties. Taken together, these changes exacerbate the tissue’s ability to withstand normal joint loading, and mechanobiological processes involving the remaining live chondrocytes cause a vicious cycle of loading-induced matrix catabolism and increased chondrocyte death within the hostile inflammatory environment associated with injured synovium and other joint tissues. Several of these issues have been reviewed in detail previously [1, 2, 18, 19]. The following sections summarize recent literature and work in progress on studying matrix damage, altered biomechanical and transport properties of injured cartilage, chondrocyte viability, and the response of injured cartilage to inflammatory mediators and mechanical loading, which is relevant to rehabilitation post-injury.
Immediate (by 1–24 h) | Acute (1 day to weeks) | Chronic (years) |
|---|---|---|
Cell necrosis | Apoptosis | Joint tissue remodeling |
GAG loss | Inflammatory mediators | Inflammation |
Collagen rupture | Leukocyte infiltration | Failed attempt at repair |
Cartilage swelling | Continued GAG loss | Arthrofibrosis |
Hemarthrosis | Matrix degradation | |
Deficient lubricants |
Damage to Cartilage Extracellular Matrix
As described above, cartilage changes at the matrix level are most often thought to begin with the immediate release of aggrecan-containing GAG constituents, macromolecules that are directly related to both the compressive [20] and shear [21] moduli of cartilage. Rapid release of GAGs at low but significant levels from mechanically injured cartilage has been reported in many studies (as reviewed in [1, 22]). During the immediate time period following injury, this GAG release is associated with purely mechanical damage to the tissue, since inhibitors of aggrecanases and matrix metalloproteinases, as well as inhibitors of biosynthesis, do not abrogate this release [23]. Over subsequent days and months in vivo, however, the release of proteolytically generated aggrecan fragments becomes more significant; these fragments are found in synovial fluid samples from ACL-injured patients and therefore have the potential to become important molecular biomarkers of the progression of PTOA [24]. This process highlights the importance of the inflammatory component of the acute and follow-on stages of joint injury and has been simulated in studies in vitro involving coculture of mechanically injured cartilage with inflammatory cytokines [25] or coculture with injured joint capsule tissue [26] (highlighting both aggrecanase activity and transcription). The soft superficial zone of immature cartilage is particularly vulnerable to compressive injury, and loss of GAG occurs predominantly from the superficial zone [27]. Loss of GAG following a single compressive impact of adult bovine cartilage increases with the rate of loading [28].
Injury can cause immediate acute damage to the collagen network as well [28], including microdamage even in the absence of overt cracks or fractures. Collagen damage is thought to be an irreversible step in the progression to PTOA [29], and such deterioration is clearly seen in the loss of tensile behavior of the tissue [30]. In addition, Maroudas [31] showed that human OA cartilage having a fibrillated collagen network would swell significantly more than normal cartilage, even after loss of a substantial percentage of GAGs: the residual swelling pressure of the remaining GAGs could not be restrained by the fibrillated collagen network. Such swelling has been observed in vitro following injurious compression of explants [32, 33], and increased levels of denatured collagen neoepitopes [34, 35] are also seen, indicative of damage to the collagen network.
While matrix changes associated with loss of GAGs and damage to collagen have immediate effects on cartilage biomechanical properties, injury to cartilage can damage and/or release many other proteins as well. For example, quantitative mass spectrometry analyses [36] showed that cartilage explants subjected to a single injurious mechanical compression released over 500 proteins (intracellular and extracellular) to the medium. Such release could result from injury-induced increases in protein degradation or enhanced synthesis. Interestingly, many of the released extracellular proteins came from the pericellular matrix (PCM), and their release could be the result of higher turnover in the PCM or increased damage to the PCM with injury. Consistent with such findings in vitro, synovial fluid samples from patients after acute injury also reveal a variety of matrix proteins [11, 37]. The composition of lubricant molecules such as hyaluronan and proteoglycan 4 are also altered after injuries (see [38] for a review). The challenge in the search for biomarkers of cartilage injury remains to identify the tissue source of such fragments in order to understand whether they come from cartilage and/or other soft tissues or bone.
Altered Biomechanical, Physicochemical, and Transport Properties of Injured Cartilage
The effects of mechanical injury on the biomechanical and biophysical properties of cartilage tissue have been reviewed extensively in previous publications that summarize findings in animal models in vivo and in cartilage explant systems in vitro (e.g., [22, 39, 40]). Here, we focus on a few specific biomechanical and conceptual issues in the context of recent findings.
The response of cartilage to mechanical impact injury seen in animal studies is exemplified in the recent report of Borelli et al. [41], who used a pendulum device to deliver a single 3 mm-wide impact to the medial condyles of 3-month old rabbits, below the fracture threshold, but estimated to be at a high ~100 MPa peak stress. Creep indentation of condyle cartilage harvested immediately and at 1 and 6 months after impact revealed an immediate decrease in injured cartilage thickness (by ~40 %), a twofold increase in equilibrium creep strain, and a significantly impaired ability for the injured cartilage to recover its thickness after creep deformation [41]. Thus, a single high impact appeared to cause damage to the collagen network, thereby resulting in a dramatic loss of the tissue’s poroelastic properties even in the absence of overt fracture. In a complementary study of the effects of repetitive abnormal injurious loading of glenoid cartilage in a rat model of rotator cuff tears, Reuther et al. [42] also reported a significant decrease in the thickness of anteroinferior region cartilage by 4 weeks. Using indentation, they performed stress relaxation tests and found a significant decrease in the equilibrium modulus calculated at 20 % indentation strain at several specified locations along the joint surface.
In general, studies in vivo can best mimic the effects of true joint anatomy and enable assessment of systemic processes, responses to injury-induced inflammatory mediators, and the effects of multi-tissue cross talk. However, variations associated with animal age, species, and geometry of loading can make it difficult to extrapolate certain findings to injury in humans [39]. Cartilage explant studies enable more precise control of injurious mechanical stimuli and the immediate cell biological environment, thereby enabling mechanistic studies of injury-induced matrix and cellular response pathways. In addition, it is possible to separate the direct effects of mechanical damage to cartilage, alone, from the combined effects of injurious loading and the subsequent presence of inflammatory mediators (simulated by adding cytokines and/or coculture with other joint tissues), which can synergistically degrade cartilage biomechanical properties with time in culture. Nevertheless, conclusions from in vitro studies must also be interpreted with caution, given the limitations of extrapolation to in vivo conditions.
For almost two decades, in vitro models have been developed to study the effects of acute mechanical trauma on articular cartilage. To help define the range of stresses, strains, and strain rates that initiate cartilage injury, Morel and Quinn [43] subjected adult bovine cartilage disks to unconfined compression at different strain rates (over 5 orders of magnitude) and peak stresses (between 3.5 and 14 MPa). The applied strain rates were above and below the tissue’s intrinsic poroelastic relaxation time, τ ~ [δ 2/(Hk)], for the tissue having equilibrium modulus H, hydraulic permeability k, and characteristic tissue distance δ through which fluid flows. Strain rates below the relaxation time τ resulted in the highest final strains; cell viability was lost throughout the tissue depth but no cracks in the matrix occurred. In contrast, high strain rates resulted in impact-like surface cracking with cell death only near the tissue surface. Consistent with these findings, Kurz et al. found a dose-dependent decrease in compressive and shear stiffness and an increase in tissue swelling, with increasing strain rate [44].
It is well known that the biomechanical properties of articular cartilage vary substantially with age, species, and joint type [45, 46]; thus, it is not surprising that the tissue’s resilience to mechanical impact injury also varies dramatically with these parameters. The soft superficial zone of immature bovine cartilage is especially vulnerable to compressive injury, causing superficial matrix disruption and extensive compaction which results in immediate and complete loss of biomechanical function compared to the deeper zone tissue of the same explants, which is much less affected [47]. In contrast, while the equilibrium modulus of the adult bovine tissue decreased in response to a “high”-impact injury, imparted by a drop tower to create grossly identifiable damage [48], the decreased stiffness was still ~55 % that of controls after 24 h, rather than the total loss of stiffness found in immature tissue [47]. In another drop tower-based blunt impact injury test using adult bovine osteochondral plugs, a decrease in creep strain in injured specimens versus control was observed by 7 days after injury, though no immediate decrease was found [49].
Micromechanical damage to cartilage matrix induced by injurious loading may also affect the transport of potential therapeutic drugs and diagnostic contrast agents into and at the surface of cartilage tissue. For example, Moeini et al. [50] observed that fluorescently tagged macromolecules showing potential for detection of surface injuries displayed decreased adsorption onto cracked surfaces of mechanically injured cartilage. These properties might aid in detecting microdamage or biochemical changes at the surface. Byun et al. found that 48 kDa anti-IL-6 Fab fragments (examined as a potential therapeutic) took over 3 days to diffuse into immature bovine and adult human femoropatellar groove cartilage explants; however, their uptake into these same explants increased significantly following compressive mechanical injury [51]. Bajpayee et al. used 66 kDa avidin protein as a model for charge-driven nanoparticle transport into cartilage and drug delivery for treating early stage post-traumatic osteoarthritis [52]. The high positive charge of avidin resulted in an uptake into normal young bovine cartilage that was 400-fold higher than that of its electrically neutral, same-sized counterpart, NeutrAvidin. Even after 40 % GAG depletion of explants to mimic early stages of PTOA cartilage degradation (i.e., similar to Fig. 11.1), avidin uptake ratio was still as high as 24 [52]. When injected intra-articularly in rats, avidin was able to penetrate the full thickness of articular cartilage within 6 h and was retained for 7 days, with a half-life of 29 h in rat cartilage [53]. The potential application of such approaches for new modes of intra-articular therapy is reviewed by Evans et al. [54].
Finally, there is still no routine, widely used approach for real-time measurement of the biomechanical properties of human cartilage during clinical examination post-injury. A recent pilot study using a handheld indentation instrument [55] was conducted to map the biomechanical properties of normal human cartilage in vivo. While the authors concluded that their approach would enable comparison measures of suspected degenerative cartilage, the instrument used could only measure force and displacement and, therefore, intrinsic material moduli could not be assessed. Kiviranta et al. [56] improved upon this instrument by incorporating ultrasound reflection measurements along with indentation; ultrasound enabled assessment of original tissue thickness from which stress, strain, and tissue dynamic modulus could be computed, as demonstrated using ex vivo human cadaver patellae.
Another recently developed instrument with the potential for post-injury arthroscopic assessment of cartilage is a streaming potential-based device [57] which was reported to be more sensitive to impact-related cartilage changes than direct biomechanical measurement. A further evolution of the use of deformation-induced streaming potentials for cartilage assessment post-injury is a completely noninvasive approach [58] in which electrodes at the surface of the knee are shown to detect streaming potentials emanating from cartilage when patients shift body weight from one leg to the other in a controlled manner. Finally, ex vivo measurements of the speed of sound (SOS) in human and porcine cartilage specimens using MRI combined with ultrasound [59] show the potential of another new noninvasive diagnostic tool, since SOS can provide an index of tissue elasticity.
Cell Viability and Cartilage Injury
Impact injury of cartilage can cause immediate cell necrosis, especially in the softer superficial zone, followed over the next days and weeks by apoptosis of neighboring populations of chondrocytes, as reviewed previously [1, 2]. In vitro studies have enabled quantitation of the mechanical loading parameters [30, 33, 60–62] and the effects of tissue maturity [63, 64] that are associated with cell death. For example, the incubator-housed instrument shown in Fig. 11.2 can apply computer-controlled compressive loads or displacements to individual or multiple geometrically defined cartilage explant disks held in specially designed autoclavable loading chambers that are mounted within the instrument [65]. Using such instruments or related load frames, investigators discovered that mechanical injury to isolated cartilage explants could lead specifically to apoptotic cell death [33, 60], with cell viability further compromised by the presence of inflammatory cytokines such as TNFα or IL-1 [66] as occurs in joint injury in vivo.
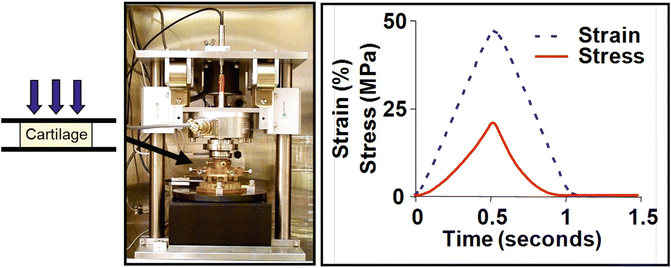
Recent studies have probed various cell- and matrix-associated mechanisms by which injurious loading may cause chondrocyte death, including injury-induced reactive oxygen species and oxidative stress [63, 67] and mitochondrial transport as a source of oxidants [68]. Imgenberg et al. [69] found that estrogen reduces mechanical injury-induced apoptosis and GAG loss in adult bovine explants. Jang et al. [70] showed that loading-induced cell death could be initiated by strain on cell adhesion receptors involving integrin-cytoskeletal interactions. Caramés et al. studied the role of autophagy, a process for turnover of intracellular organelles and molecules that protects cells during stress response [71]. They discovered that mechanical injury can suppress autophagy regulators, and pharmacological activation of autophagy, e.g., using rapamycin, can prevent cell death and GAG loss in mechanically injured explants [66].
Clinical detection of intratissue cell death soon after traumatic joint injury could aid in treatment decisions but is technically challenging; however, new approaches are on the horizon. Using equine osteochondral blocks ex vivo, Novakofski et al. showed that quantitative multiphoton microscopy can detect cell death in situ in live tissue, with clinical potential for detection of early cartilage damage [72]. In addition, Rolauffs et al. [73, 74] identified a distinct spatial reorganization of superficial human chondrocytes associated with proliferative remodeling in response to early OA lesions; these cell patterns also have potential diagnostic utility. Both approaches above have the potential to be configured for arthroscopic examination.
Cartilage Changes Post-injury in an Inflammatory Environment
Cartilage degeneration in PTOA is driven by the entire synovial joint; however, chondrocytes can play a primary role when stimulated to increase local production of matrix-degrading proteases, downregulate the synthesis of ECM molecules, and produce inflammatory mediators. In response to joint injury or cytokine stimulation, latent or newly secreted aggrecanases produced by multiple joint tissues may rapidly degrade aggrecan, significantly altering the mechanical properties of the tissue. Irreversible protease-induced collagen degradation often occurs after aggrecan depletion, suggesting that aggrecan may protect collagen fibrils from proteolytic degradation [75]. The array of known matrix proteases can also degrade many other ECM macromolecules that are responsible for homeostatic assembly and remodeling of cartilage matrix [76].
Preclinical animal models are playing an increasingly important role in the study of mechanistic pathways that regulate cartilage degradation and attempts at repair in a post-injury inflammatory environment. Over 135 strains of genetically engineered mice are now available to explore various aspects of OA, and both surgical and mechanical methods for inducing joint injury in mice can be used [77]. Animal models can highlight the importance of inflammation and multi-tissue interactions within an injured joint. For example, a recent study of ACL transection in minipigs demonstrated that MMP-13 and ADAMTS-4 were highly upregulated in the synovium and ligament and could thereby affect cartilage matrix [78]. Similarly, in an ovine model of simulated ACL reconstructive surgery, changes associated with early PTOA were associated with acute post-injury synovial inflammation [79].
At the same time, quantitative cell biological and mechanobiological pathways can be difficult to delineate in animal models, and complementary in vitro studies can add powerful approaches to the understanding of these mechanisms. Injurious compression of cartilage explants alone, even in the absence of other joint tissues, significantly increases chondrocyte gene expression levels of MMP-1, MMP-3, MMP-13, and ADAMTS-5 within 24 h after injury [80]. Cartilage explantation also activates intracellular inflammatory signaling pathways (e.g., p38, JNK, and ERK) and induces mRNA expression of IL-1α and IL-1β [81]. Biologically active IL-1 proteins were also detected in cartilage lysates [81]. In a microarray analysis of cartilage explants subjected to recutting, strong upregulation of the Wnt-16 gene was detected [82]. Wnt pathways play an important role in chondrocyte differentiation [83] and dysregulation of Wnt pathways in adult tissues could contribute to chondrocyte hypertrophy seen in OA [84]. Taken together, these results suggest that impact injury of cartilage, alone, leads to dysregulation of chondrocyte metabolism and kick-starts catabolic and inflammatory processes in cartilage.
Anabolic activities are also upregulated in cartilage subjected to mechanical injury. Gene expression levels of TIMP-1 increased by 12-fold within 24 h of explant impact injury, suggesting attempts at early repair by inhibiting matrix proteinases; in addition, activation of BMP and FGF pathways was also shown in cartilage following mechanical injury [85, 86], which may play a role in reparative responses of cartilage to injury. These increases in anabolism appear to be attempts to offset the catabolic effects of MMPs, aggrecanases, and catabolic cytokines and chemokines. However, attempts to increase matrix synthesis are compromised by the presence of inflammatory responses in the synovial joint.
Traumatic joint injury rarely involves disruption of cartilage surface alone and may include bone bruises, rupture of ligaments and menisci, and lesions in the joint capsule and synovium. In vitro models have been developed to include aspects of these multi-tissue injuries with associated inflammation. These models are based on the knowledge that elevated levels of IL-1β, TNFα, IL-6, IL-8, and other inflammatory mediators are found in synovial fluid of patients with acute knee injuries [11], and concentrations of such mediators correlate with severity of damage [87]. Among the proinflammatory cytokines, IL-1 and TNFα are considered the major players. Several studies have shown that IL-1 and TNFα promote cartilage matrix degradation by inducing expression of extracellular matrix-degrading enzymes (MMP-1, MMP-3, MMP-13, ADAMTS-5) [88], inhibiting collagen and aggrecan synthesis [89], inhibiting anabolic activity of growth factors, and inducing production of IL-6 [90] and chemokines such as IL-8 [91]. The role of inflammatory cytokines in the pathogenesis of OA has been reviewed in more detail by Kapoor et al. [92].
Based on this knowledge, in vitro models have incorporated coculture of mechanically injured immature bovine and adult human cartilage explants with specific inflammatory cytokines such as IL-1 alone [93], TNFα alone [93], or the combination of TNFα, IL-6, and the IL-6 soluble receptor sIL-6R [25]. (The latter choice was motivated by the previous finding that IL-6 with its soluble receptor augments proteoglycan catabolism from cartilage [94].) These studies showed that mechanical injury potentiates aggrecan catabolism induced by inflammatory cytokines (Fig. 11.3). The value of such in vitro models for use in the discovery of potential therapeutics has been demonstrated by the subsequent discovery that dexamethasone (Dex) essentially blocks the catabolic effects of injury plus cytokine treatment on GAG loss (Fig. 11.3a) [95] while preserving cell viability and metabolism. Even after incubation of explants with IL-1, which caused almost total GAG loss by 12 days, addition of Dex could completely inhibit collagen degradation (Fig. 11.4) [96].
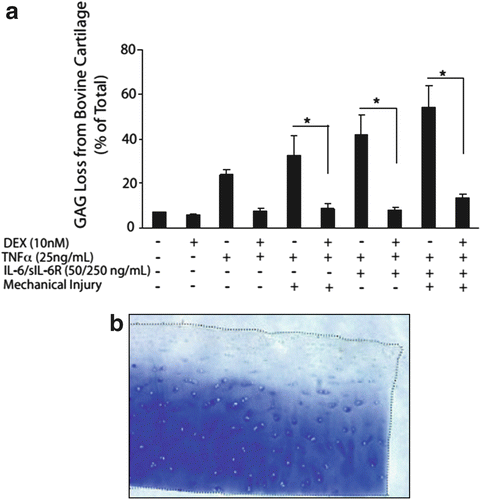
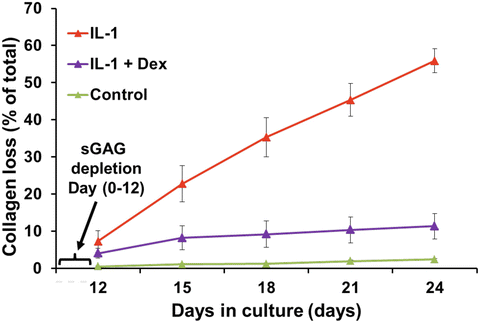
Fig. 11.3
(a) Effects of dexamethasone on sGAG loss in young bovine cartilage treated with combinations of mechanical injury, TNFα, and IL-6/sIL-6R for 6 days. The injury was applied using the waveform of Fig. 11.2. Dex abrogated sGAG loss caused by injury plus cytokine treatment in this system (from [95]). Similar trends were found with adult human knee cartilage explants. (b) Toluidine blue staining of 0.8-mm disks of human knee cartilage with intact superficial zone treated with injury plus TNFα plus IL-6/sIL-6R over 4 days (from [25]); the spatial profile of GAG loss and histological appearance of the tissue is similar to that of the human clinical biopsy specimen of Fig. 11.1
Fig. 11.4
Bovine cartilage explants were treated with 1 ng/ml IL-1 continuously for 24 days. After almost complete loss of sGAG by day 12, dexamethasone was added to the cultures, and collagen loss was measured. Dexamethasone was able to prevent proteolytic degradation and loss of collagen (from [96])
In parallel complementary studies, coculture of normal or mechanically injured cartilage explants with injured (explanted) joint capsule specimens has been used to test the broader hypothesis that multiple factors from the synovium could induce catabolic pathways within neighboring cartilage. Patwari et al. [97] observed that coincubation of human joint capsule tissue with normal human knee cartilage explants inhibited chondrocyte biosynthesis through an IL-1-independent signaling pathway. Using this model, Lee et al. [26] studied the temporal evolution of 21 genes (by qPCR) in normal or injured cartilage cocultured with joint capsule explants; clustering analyses enabled identification of co-expression profiles for genes associated with injury alone, coculture alone, or injury plus coculture. While MMP-13 and ADAMTS-4 clustered with the effects of coculture, ADAMTS-5 expression and activity (by immunohistochemistry) clustered with injury and injury plus coculture.
Related posts:
 Arthritis That Develops After Joint Injury: Is It Post-Traumatic Arthritis or Post-Traumatic Osteoarthritis?
Post-Traumatic Arthritis: Definitions and Burden of Disease
Potential Targets for Pharmacologic Therapies for Prevention of PTA
Arthritis That Develops After Joint Injury: Is It Post-Traumatic Arthritis or Post-Traumatic Osteoarthritis?
Post-Traumatic Arthritis: Definitions and Burden of Disease
Potential Targets for Pharmacologic Therapies for Prevention of PTA
 Outcomes of ACL Injury: The MOON Consortium
Outcomes of ACL Injury: The MOON Consortium
 Biomarkers of PTA
Biomarkers of PTA
 Instability: Dynamic Loading Models
Instability: Dynamic Loading Models
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


