Fig. 6.1
Bar graph showing the association between the changes in NAA and ATP cerebral concentrations as a function of the severity of TBI. Rats underwent different traumatic insults (mild, severe, severe + hypoxia-hypotension) using the weight drop model with diffuse axonal injury. Animals were sacrificed 48 h after TBI and metabolites of interest were assayed by HPLC on deproteinized whole brain homogenates. Each histogram is the mean of six animals. Standard deviations are represented by vertical bars. NAA and ATP in severe and severe + hypoxia-hypotension rats were different from either values recorded in controls (p < 0.01) or from those recorded in mild-injured animals (p < 0.01)
In this study, an impressive similarity in the time-course modifications in these two compounds after graded injury was observed and it was shown that 48 h post-injury was the minimal time point at which one can clearly distinguish the severity of TBI, from a metabolic point of view [54]. These bench data strongly supported the indication for a potential role of NAA in quantifying neuronal damage and predicting neuropsychological outcome after TBI and being of high clinical relevance since the use of 1H-MRS allows for the measurement of NAA noninvasively in vivo [57–60]. In addition, the metabolic recovery observed in the mTBI animals implied that the process leading to the reversible NAA and ATP reductions was attributable to transient biochemical changes and not simply to cell death. Beyond showing the profound TBI-induced modification in NAA homeostasis, this finding clearly demonstrated that different levels of “physical” injury correlated with different levels and kinetics of “biochemical” damage, which are reversible in mTBI and irreversible in severe TBI (sTBI) [37, 54].
Substantial evidence indicates that NAA synthesis takes place exclusively in neurons, that it is strictly tied to neuronal energy metabolism and mitochondrial functions, and that the distribution pattern of NAA closely parallels the distribution of “respiratory activity” [41–46]. It has been clearly evidenced that in the general posttraumatic metabolic derangement, acetyl-CoA homeostasis is affected by graded head injury, following a pattern very similar to those observed for both ATP and NAA [61]. Therefore, in metabolic conditions of low ATP availability, when all of the pathways and cycles devoted to energy supply are operating at their maximal activity with the aim of replenishing ATP levels, acetyl-CoA will not be accessible for NAA synthesis. Only when the ATP deficiency is fully restored will acetyl-CoA become available again to be shifted into the NAA “production” pathway.
With this in mind, a decrease in NAA concentration can be seen as an indirect marker of posttraumatic metabolic energy impairment. According to these concepts, it is evident that if NAA is still below the physiological values after an mTBI, the concussive biochemical derangement involving more complex pathways than simple NAA homeostasis [34–37] cannot be considered to be resolved. Thus, NAA embodies a biochemical surrogate marker to monitor the overall cerebral metabolic status, and it appears that under conditions of decreased NAA, the cells are functional yet are still experiencing energetic imbalance. It is important to emphasize that these results on brain energy metabolism following an mTBI are consequences of the recently discovered gene program of neuroprotection discussed above [35].
Post-concussion Brain Vulnerability
The basic pathophysiological paths explored thus far have clarified some aspects of this particular clinical entity, suggesting that albeit concussion is considered a form of mTBI, should not to be considered so “mild.” With the exception of the almost always punctual reversibility of all the metabolic and molecular modifications induced, it is probably not prudent to use the adjective “mild” when referring to a traumatic event that can have so many consequences involving fundamental functions of neuronal cell metabolism. However, while all of these biochemical modifications are scientifically interesting, they might appear, at a first glance, of negligible clinical utility because they are all spontaneously and fully reversible; moreover, patients with mTBI show no or minimal focal neurological problems and generally show a “radiologically” normal brain.
Despite the apparently “calm” clinical scenario and the reversible biochemical/metabolic damage, a reasonable body of evidence clearly demonstrated that “concussed” brain cells, although still perfectly functioning, during this period of molecular and biochemical alterations are truly under a peculiar state of “vulnerability,” originally proposed by Hovda and colleagues [62]. If during this limited period of time neuronal cells sustain a second, typically nonlethal, insult in a close temporal proximity from the first, they would suffer irreversible damage and die [62]. Subsequent studies have strongly corroborated these original findings showing that during this transient period of altered brain metabolism, a second concussive episode may cause significant additional neuronal damage [63, 64]. In other words, concussion induces a pathophysiologic condition, mainly manifested by mitochondrial malfunctioning and energetic metabolic perturbations, that makes the brain more susceptible to a second trauma, even if of modest entity. This occurrence is capable of causing severe and sometimes irreversible cellular injury, thereby creating a disproportion between the trauma severity and the subsequent cerebral damage. It is worth recalling that, in the clinical setting, the possibility of having a second concussive injury within a not yet defined period of time from the first (i.e., days or weeks) has been reported to be even fatal in some instances [65–69], a phenomenon also known as the second impact syndrome (SIS), occasionally encountered in sports medicine [70].
Several studies in animals in which investigators focused on mTBI-induced dysfunction have been published, and current data support the concept of transient biochemical and physiologic alterations that may be exacerbated by repeated mild injuries within specific time windows of vulnerability [61, 63, 64]. In a rat weight drop experiment performed by applying a new and easily reproducible protocol to simulate a “second impact” condition, it was clearly demonstrated that levels of NAA, ATP, and the ATP/ADP ratio decreased significantly as a function of the time interval between two repeat concussions [61]. Maximal metabolic abnormalities were seen when the occurrence of two mild injuries was separated by a 3-day interval; in fact, the metabolic abnormalities in these animals were similar to those occurring after a single sTBI. However, when the two concussions were spaced by 5 days, results of the biochemical parameters were identical to those recorded in rats experiencing a single episode of mTBI (see also Fig. 6.2).
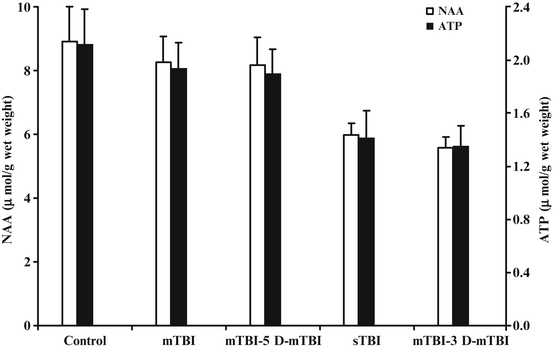
Fig. 6.2
Bar graph showing the changes in NAA and ATP cerebral concentrations in rats subjected to single mild or sTBI, or to repeat mTBI spaced by different time intervals (3 or 5 days). The traumatic insults were induced using the weight drop model with diffuse axonal injury. Animals were sacrificed 48 h after the last TBI and metabolites of interest were assayed by HPLC on deproteinized whole brain homogenates. Each histogram is the mean of six animals. Standard deviations are represented by vertical bars. No differences in both NAA and ATP were observed between a single mTBI and a repeat mTBI with 5-day interval (also no different from controls), as well as between a single sTBI and a repeat mTBI with 3-day interval (metabolites in these two groups of animals were significantly different from those recorded in controls, p < 0.01). This indicates that a second mTBI has no cumulative effects with the first one when falling outside the window of brain vulnerability; otherwise, the effects on brain metabolism are those observed after a single sTBI (cumulative effects in the case of two repeat mTBIs spaced by 3 days or more specifically the second injury occurs inside the window of brain vulnerability)
The other astonishing observation about this study was that of an identical 10 % mortality rate in animals subjected to a single sTBI and animals doubly injured by mTBI with a 3-day trauma interval; on the contrary, no animals died when subjected to a single mTBI or double-impact mTBI with a 5-day interval [61]. In a follow-up study, similar perturbations were found to persist as late as 7 days after double impact in animals receiving two mTBIs spaced by 3 days, indicating cumulative effects from repeat mTBI caused irreversible modifications of brain metabolism equal to those provoked by a single sTBI. Similar data were reported by Laurer et al. [71] in a histopathology study in which they described the important cumulative effects of two episodes of mTBI (24 h apart) in mice, which led to pronounced cellular damage when compared with animals that sustained a single trauma only. The authors concluded that although the brain was not morphologically damaged after a single concussive insult, its vulnerability to a second concussive impact was dangerously increased [71]. These results were reproduced in additional studies, either in the juvenile [72] or in the adult rat [73] thus corroborating the initial findings suggesting that the metabolic alterations can persist for days after concussion, creating no morphological damage but representing the pathological basis of the brain’s vulnerability [62, 74].
According to the results of these studies, it may be affirmed that if the second concussion finds the cells fully committed to recover from the first insult, but still in a perfectly reversible energetic failure, it will cause further mitochondrial malfunctioning, leading to the same irreversible metabolic damage (characterized by permanent energetic failure) observed after a single severe injury [61, 73]. Thus, two mTBIs that occur too close in temporal proximity can replicate the effects of a single sTBI. The key biochemical issue of the vulnerable brain lies in the incomplete resolution of the initially reversible energetic crisis triggered by the first insult and, very probably, by the profound changes in the pattern of gene expression aimed at neuroprotection [35]. The foremost clinical implication of these experimental data is that within days after injury, the metabolic effects of two concussions can be dangerously additive. This information might not be surprising; however, similar human data regarding brain metabolites currently are not available. The second clinical implication of this notion is again remarkable because it is very difficult to establish how long the aforementioned period of brain vulnerability will last and when the occurrence of a second trauma would be uneventful.
In summary, all these preclinical data provide the experimental demonstration of the exquisitely metabolic nature of “brain vulnerability” kicked off by an mTBI, and offer a unique contribution to the complex biochemical damage underlying the clinical scenario of repeated concussive trauma, sometimes leading to catastrophic brain injury. Moreover, these data highlight that concussion triggers changes in cerebral cell homeostasis that can be evaluated at the molecular level, therefore representing key parameters with which to monitor the recovery of cell functions in post-concussed subjects.
“Imaging” the Brain’s Biochemistry: The Role of Proton Magnetic Resonance Spectroscopy (1H-MRS)
The above-described alterations of central energy metabolism might well represent a reasonable explanation of the striking and unpredictable discordance often observed between the minimal findings on conventional imaging [75] and the extent of neurocognitive deficits exhibited by patients with mTBI [76, 77]. As a fact, none of the currently available diagnostic tests [77–80] is capable of measuring this unique, transient, and potentially dangerous state of metabolic vulnerability of the “concussed” brain tissue, since changes during such a period are “visible” at the molecular level only.
The relevance of NAA as a biochemical marker of the metabolic neuronal “wealth” brings in a valid possibility to investigate changes in brain metabolism, stating that NAA and other groups of metabolites can be noninvasively measured “in vivo” by 1H-MRS. This technique is based on the ability to localize the MRS signal into a specific volume of tissue, therefore providing a real-time “image” of the neurochemistry of certain brain regions, which in pathological states, differs from healthy tissue. The hardware for MRS is similar to standard MRI, requiring only additional software to suppress the signal of water, to capture the signal of various compounds at variable wavelength, and to provide analysis and imaging. Data can be obtained from either a single volume of brain, known as the single voxel spectroscopy, or a 2- or 3D analysis simultaneously acquired over a wider region, known as the multi-voxel spectroscopy or chemical shift imaging (CSI). Briefly, when subtracting the spectral signal of the 1H of water (the most abundant molecule containing an atom with an unpaired proton, which is a prerequisite to allow to detect a molecule by MRS), the 1H-NAA resonance returns one of the best defined peaks at approximately 2 ppm. By using the clinically safe magnetic fields of the MR apparatuses (1.5 or 3.0 T) in the range from 1.8 to 3.4 ppm two additional, well-resolved, resonance peaks are obtained, one at approximately 3.2 ppm referring to “choline” (Cho) and the other at approximately 2.9 ppm referring to “creatine” (Cr). It is worth recalling that the relatively low magnetic fields of 1.53.T currently used (with no clinical side effects) do not allow to discriminate the NAAG signal in the NAA peak or the creatine phosphate (CrP) signal in the Cr peak or to resolve about the ten different compounds containing the choline moiety in their molecule in the Cho peak [53].
However, when referring to NAA and Cr, this is of limited relevance since, in the brain tissue, NAA and Cr are about ten times more concentrated than the respective related compounds NAAG [51, 56] and CrP [81, 82] and the areas of these two peaks can generally be considered as legitimate measurements of NAA and Cr. Since the Cho peak is mainly composed by phosphocholine, glycerophosphocholine, and phosphatidylcholine, quantification of this peak area is not in direct correlation with one compound only. In summary, in a typical 1H-MRS spectrum, the NAA peak represents the NAA-containing compounds (with NAA contributing for 90 % to the peak signal) and it is used as a marker of energy state and neuronal integrity, and the Cr peak represents creatine-containing compounds (with creatine accounting for 90 % of the intensity signal) and thought to be a further marker of cellular energy, while the Cho peak represents the choline-containing compounds (with phosphocholine, glycerophosphocholine, and phosphatidylcholine accounting for about 90 % of the intensity peak signal) the biological meaning of which is that of being related to the cell membrane turnover/density.
Unless determining the water content within the voxel(s) for a calculation of the absolute concentrations of the different metabolites [53, 83], it is always necessary to quantify at the same time all the three peak areas and carry out a semiquantitative evaluation of these metabolites expressed by the NAA/Cr and NAA/Cho and Cho/Cr ratios [84–89].
In a pilot study carried out in a cohort of ten concussed athletes, examined by 1H-MRS for their NAA cerebral content at different time points after the concussive event, it was demonstrated that there was a significant decrease in NAA (either in terms of the NAA/Cr or NAA/Cho ratios) during the first 15 days post-injury with a subsequent normalization occurring 30 days after concussion [90]. An important finding of this study was that all athletes declared to be symptom-free within the first 15 days following the traumatic insult when brain metabolism was far to get normalized, i.e., disappearance of symptoms and metabolism rescue did not correlate. Furthermore, notwithstanding it was mandatory to restrain from physical activity before normalization of brain metabolism, three additional athletes enrolled in this study, prompted by the clearance from post-concussive symptoms, returned to play and experienced a second concussion between the tenth and the thirteenth day after the first insult. Although they were not affected by SIS nor showed signs of sTBI, they showed a significant delay in NAA normalization.
The results of this pilot study were confirmed by a recent multicenter clinical trial involving 40 concussed athletes and 30 healthy volunteers [91]. Thanks to the involvement of three neuroradiologic centers, we were initially able to demonstrate that, despite different combinations of field strengths (1.5 or 3.0 T) and modes of spectrum acquisition (single- or multi-voxel), results on relative NAA quantification did not depend on the scanners currently in use in most neuroradiology centers, confirming that NAA determination represents a quick (15-min), easy-to-perform, noninvasive tool to accurately measure changes in cerebral biochemical damage occurring after a concussion. The cohort of 40 concussed athletes was evaluated at 3, 15, 22, and 30 days post-impact by 1H-MRS for the evaluation of brain metabolism. Patients exhibited the most significant decrease in NAA (evaluated in terms of metabolite ratios) at day 3 post-injury, showing a gradual nonlinear recovering, initially slow and, following day 15, faster.
At 30 days post-injury, all subjects showed complete recovery, having metabolite ratios not different from values detected in controls (see also Fig. 6.3). It is very important to underline that all these 40 patients self-declared symptom clearance between 3 and 15 days after concussion, confirming results of the pilot study [90] and strongly demonstrating differential times of disappearance of clinical gross signs and of normalization of NAA levels.
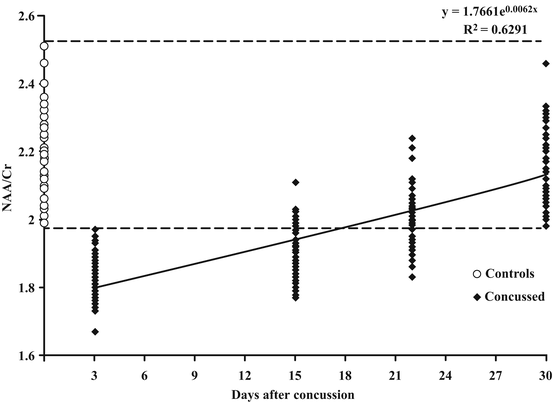
Fig. 6.3
Time-course changes in the NAA/Cr ratio recorded in 40 concussed athletes using proton magnetic resonance spectroscopy (1H-MRS). Control values of the NAA/Cr ratio were obtained in a group of 30 age- and sex-matched healthy subjects, having no prior history of concussions. Recovery of brain metabolism followed a nonlinear trend, according to the exponential equation reported in the figure and suggested a slow recovery in the first 15 days and a much faster recovery in the second 15 days. In this group of athletes, 30 days was the time necessary to have the NAA/Cr ratio in all subjects fall within the range of variability of controls. It is very important to note that ALL athletes declared to be symptom-free at the time of the second 1H-MRS when only 5/40 had values of the NAA/Cr ratio similar to those of controls, concluding that there was no correlation between clinical healing and normalization of brain metabolism
As clearly indicated in Fig. 6.4, it is also worth mentioning that the individual rate of NAA recovery varied from athlete to athlete, thus suggesting that this variation might have been due to possible differences in the severity of each concussion.
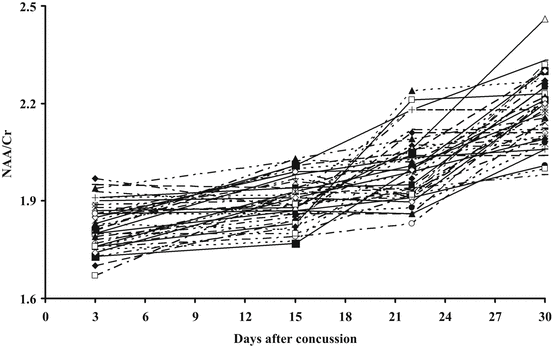
Fig. 6.4
 Neuropsychological Testing in Sports Concussion Management: An Evidence-Based Model when Baseline Is Unavailable
Metabolic Dysfunction Following Traumatic Brain Injury
Biomarkers for Concussion
Current Understanding of Concussion: Treatment Perspectives
Neuropsychological Testing in Sports Concussion Management: An Evidence-Based Model when Baseline Is Unavailable
Metabolic Dysfunction Following Traumatic Brain Injury
Biomarkers for Concussion
Current Understanding of Concussion: Treatment Perspectives
 Acute and Lingering Impairments in Post-concussion Postural Control
Acute and Lingering Impairments in Post-concussion Postural Control
 Advanced Neuroimaging of Mild Traumatic Brain Injury
Advanced Neuroimaging of Mild Traumatic Brain Injury




Individual recovery of brain metabolism, evaluated in terms of NAA/Cr ratio by 1H-MRS, occurring in the 40 concussed athletes shown in Fig. 6.3. As it is evident from this re-plotting, the rate of recovery, the time needed to restore normal concentration of metabolites related to brain energy metabolism, varies from subject to subject thus meaning that full normalization may occasionally be obtained earlier than 30 days after injury
Related posts:
Neuropsychological Testing in Sports Concussion Management: An Evidence-Based Model when Baseline Is Unavailable
Metabolic Dysfunction Following Traumatic Brain Injury
Biomarkers for Concussion
Current Understanding of Concussion: Treatment Perspectives
Acute and Lingering Impairments in Post-concussion Postural Control
Advanced Neuroimaging of Mild Traumatic Brain Injury
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
