Systemic Diseases Resulting in Hip Pathology
Colin T. Penrose
John W. Barrington
The vast majority of hip replacements are performed as a result of degenerative conditions, such as osteoarthritis, idiopathic avascular necrosis, or arthritis secondary to dysplasia or trauma, as well as rheumatologic conditions such as rheumatoid arthritis. However, there are several other rheumatologic as well as hematologic, metabolic, endocrine, and congenital conditions that can result in sufficient hip joint damage to require surgical intervention. Although total hip arthroplasty (THA) has proven to be an effective treatment for the vast majority of conditions that result in loss of hip joint cartilage, the surgeon treating patients who have systemic illness involving the hip joint must be cognizant of specific features that may affect clinical and treatment decisions. Such features may affect the patient’s ability to tolerate the surgery required, they may alter the technical requirements of the surgery in specific ways, and they may alter the nontypical natural history of the implanted hip arthroplasty in a specific cohort of patients. This chapter reviews several of the most common conditions with which the hip surgeon should be familiar.
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a disease in which organs are damaged by pathogenic autoantibodies and immune complexes. Although all ages and both sexes can be affected, the majority of the cases are in women of child-bearing age. A study of 395 Chinese patients with SLE concludes that childhood onset of the disease increases the risk of low bone-mineral density (1). The arthritis associated with SLE usually involves the hand and wrist, but any joint is susceptible. Endstage disease in the hip, however, is almost always secondary to avascular necrosis in patients taking corticosteroids. Several studies have documented corticosteroid-associated osteonecrosis (2,3,4,5) and several approaches have been tested to improve outcomes: Prasterone to protect against low bone-mineral density (6), calcium or calcitrol (7) or ibandronate to increase cortical bone density (8), extracorporeal shock-wave therapy (9), transtrochanteric anterior rotational osteotomy (10), and free vascularized fibular grafting (11). Symptomatic osteonecrosis occurred in 13% of one cohort of 744 patients with SLE (12), and often involved multiple joints. Survival in patients with SLE is approximately 70% over 10 years, with infection and renal failure being the leading causes of death (13).
In one report of 43 prosthetic hip replacements in 31 patients with SLE (14), all but 4 patients had osteonecrosis: 1 patient had degenerative disease and 3 others had displaced femoral neck fractures. There were 29 THAs performed, and all but 3 were rated good or excellent at a mean follow-up time of 66 months. Complications included delayed wound healing in 15% and superficial wound infection in 10%. Although no patients who had a THA required revision, 5 of 14 bipolar arthroplasties required revision. Only five patients (seven hips) with bipolar arthroplasties had a good or excellent result, and there was a high incidence of persistent groin pain in this group of patients. Of these patients, 25% died less than 5 years after the operation from disease-related complications. Based on the results (published in 1987), these authors recommended cemented THA for this group of patients.
In another report, 26 hip arthroplasties (15 uncemented, 11 other) in 19 patients were followed for a mean of 4.6 years and compared with a matched control group (15). The visual analog pain scores, Harris hip scores, and SF-36 scores were similar in the two groups. In the SLE group, the complications included small calcar cracks in four hips, two early dislocations, one persistent thigh pain, and one asymptomatic cup loosening. No other symptomatic osteolysis or loosening was observed.
Sickle-Cell Hemoglobinopathies
Osteonecrosis of the femoral head is a frequent and important complication of the sickle-cell hemoglobinopathies, which include sickle-cell anemia (hemoglobin S-S genotype), hemoglobin SC disease, hemoglobin S–β-δ-thalassemia disease, and hemoglobin S–β+-thalassemia disease. Concomitant α–thalassemia, which may modify the course of the disease, affects about 30% of patients with sickle-cell anemia (16).
Prevalence
In a multicenter study of 2,590 patients with a sickle-cell hemoglobinopathy, the prevalence of osteonecrosis of one or both femoral heads was 9.8% (253 patients) (16). During this 3-year study, the prevalence was 13% in the S–β-δ-thalassemia group of patients, 10% in the hemoglobin S-S group, 8.8% in the hemoglobin S-C group, and 5.8% in the S–β+-thalassemia group. The prevalence of osteonecrosis was 8.9%, with the highest incidence among patients with the hemoglobin S-S genotype and α-thalassemia. The median age at diagnosis was 28 years for patients with the hemoglobin S-S genotype and α-thalassemia, 36 years for those with hemoglobin S-S genotype without α-thalassemia, and 40 years for those with hemoglobin S-C genotype. Bilateral involvement of the femoral head occurs frequently in sickle-cell hemoglobinopathies, and it developed in 54% of patients involved in this study.
The natural history of osteonecrosis in sickle-cell disease is one of clinical and radiographic progression. A study from one center in France found that of 75 hips in adults without collapse (Steinberg stage I and II) at initial presentation, 65 (87%) demonstrated collapse within 5 years of diagnosis, with an average time between diagnosis and collapse of 42 months for stage I hips, and 30 months for stage II hips (17). A more recent report from this same center found that asymptomatic osteonecrosis, which presents when the contralateral hip is symptomatic, is highly likely to progress to pain (91%) and collapse (77%) in a series of 121 hips followed for 10 to 20 years with a mean of 14 years (18). Of the 56 patients initially given a Steinberg grade of 0 in their asymptomatic hip, 84% were symptomatic and 61% had collapse during the most recent follow-up (on average 14 years later). Of the 42 hips originally asymptomatic initially deemed stage I, 95% of the patients experienced pain and 86% had femoral head collapse within only 3 years. All 23 patients with asymptomatic hips initially considered stage II progressed to pain and collapse within 2 years. In a separate report from this same center, with 15-year average follow-up, 80% of hips with the onset of osteonecrosis during childhood were painful and had decreased mobility, limb-length discrepancy, or an abnormal gait (19).
Pathophysiology
Osteonecrosis in sickle-cell hemoglobinopathies is likely initiated by localized sludging of sickled cells in the marrow sinusoids, leading to necrosis of bone marrow and osteocytes. The subsequent repair process may lead to healing, especially in younger patients, but it may also produce increased intramedullary pressure and ultimately bone resorption, subchondral fracture, and collapse of the femoral head.
Patients with sickle-cell anemia can also have crises, which are often associated with severe polyarthralgia, and occasionally with joint effusion and synovitis. Joint effusion and synovitis result from infarction of the synovium. Rarely, patients with sickle-cell anemia present with an inflammatory arthritis of the hip, with protrusio acetabuli, similar to that seen in patients with rheumatoid arthritis.
Other Organ-System Involvement
Osteonecrosis of the femoral head in the sickle-cell hemoglobinopathies is frequently associated with other chronic organ-system disease. These patients may develop retinopathy, chronic renal disease, cerebrovascular accidents, acute or chronic pulmonary disease (acute chest syndrome), congestive heart failure, deep vein thrombosis, gallstones, and splenic infarcts. Patients with sickle-cell disease are susceptible to Salmonella osteomyelitis because of a defect in the complement pathway. The increased rate of infection that has been reported after THA is multifactorial and may be related to functional asplenia, an abnormal immune system, and abnormal intraosseous perfusion. Patients also develop chronic skin ulcers in the lower legs and sloughing of the intestinal mucosa, which lead to recurrent episodes of bacteremia and late hematogenously induced prosthetic hip infection.
Life Expectancy
In considering surgical intervention in patients with sickle-cell hemoglobinopathy, it is important to consider the life expectancy and risk factors for early death. In a multicenter study of 3,764 patients, the median age at death was 42 years for men and 48 years for women (20). However, 50% of patients with sickle-cell anemia survived past the fifth decade of life. Among patients with hemoglobin S-C disease, the median age at death was 60 years for men and 68 years for women. Patients with renal failure, acute chest syndrome, seizures, and a low level of fetal hemoglobin were associated with an increased risk of early death.
Imaging
In most studies of osteonecrosis of the femoral head in sickle-cell hemoglobinopathies, femoral head osteonecrosis is staged according to the classification of Ficat as described on plain radiographs. In the multicenter study of 2,590 patients mentioned previously, osteonecrosis developed in a subgroup of 135 patients, of whom 47% had stage II disease (sclerosis and radiolucent areas), 30% had stage III disease (subchondral lucent line or collapse), and 23% had stage IV disease (late changes) (16). In this study, almost half the patients (47%) in whom osteonecrosis developed had no pain in the hip or limitation of motion at the time of diagnosis. However, one-fifth of these patients developed symptoms later. Magnetic resonance imaging (MRI) can detect stage I disease in patients with sickle-cell hemoglobinopathies (Fig. 36.1) and is recommended when a patient presents with pain in the hip, thigh, or knee, despite normal radiographs.
Importance of Disease Management
Early identification and management of hip pain in patients with sickle-cell hemoglobinopathies can improve their outcomes by reducing the vaso-occlusive crises thought to cause femoral head avascular necrosis. A study of 215 patients (94 with SS and 121 SC) suggests the importance of comprehensive orthopedic monitoring and systematic care (21). This study found that using hydration, rest, crutches, and pharmacologic pain management to treat and prevent the painful crises
of the joint associated with this disease could delay or even prevent the onset of femoral head avascular necrosis. Relative to their control series of 115 patients (58 SS and 57 SC), the comprehensive disease management group had a reduction in femoral head avascular necrosis (36.5% compared to 14.4%).
of the joint associated with this disease could delay or even prevent the onset of femoral head avascular necrosis. Relative to their control series of 115 patients (58 SS and 57 SC), the comprehensive disease management group had a reduction in femoral head avascular necrosis (36.5% compared to 14.4%).
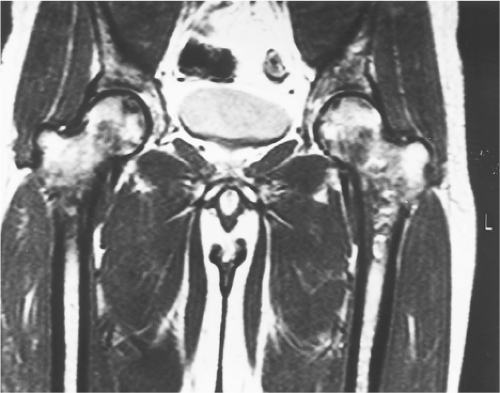 Figure 36.1. An MRI scan of a 32-year-old man with sickle-cell β-thalassemia hemoglobinopathy and left hip pain confirms the diagnosis of osteonecrosis of both hips. |
Preoperative Preparation
Complications after operative procedures on the hip in patients with sickle-cell anemia are common, and the rate of postoperative complications has been reported to be as high as 50%. Thus, a thorough preoperative medical and hematologic evaluation is required prior to major hip surgery, especially THA. The operation should be delayed if the patient has any pulmonary problem or leg ulcers.
To decrease the incidence of complications, preoperative transfusion to a hemoglobin level of 10 g/dL, with or without exchange transfusion to decrease the level of hemoglobin S to less than 30%, is generally recommended. In a multicenter, randomized study of transfusion regimens in 692 surgical procedures (72 orthopedic procedures), and in a follow-up study which included 66 additional orthopedic procedures, it was found that a conservative transfusion regimen (increasing the hemoglobin level to 10 g/dL) was as effective as an aggressive regimen (decreasing the hemoglobin S level to less than 30%) in preventing perioperative complications (22,23). There were only half as many transfusion-related complications in the conservative transfusion group.
For patients undergoing major hip surgery, including THA, spinal or epidural anesthesia is recommended to decrease blood loss and lower the incidence of thromboembolism. Good oxygenation and hydration throughout the hospitalization is recommended, along with use of a prophylactic antibiotic and some type of thromboembolism prophylaxis. The authors routinely utilize bilateral thigh-high pneumatic compression devices intraoperatively and after THA in patients with sickle-cell hemoglobinopathies.
Another important preoperative consideration is septic arthritis. A retrospective study of 24 patients with osteonecrotic hip joints and septic arthritis highlights the importance of hip aspirations to test for infection prior to THA in patients with sickle-cell disease and osteonecrosis (24).
Surgical Treatment (Figs. 36.2 and 36.3)
Core Decompression
Core decompression has been suggested for the treatment of Ficat stage I (normal radiograph, abnormal MRI scan) and early Ficat stage II idiopathic osteonecrosis of the femoral head. However, few reports of this procedure have included patients with sickle-cell disease. It has been suggested that the relatively young age of patients with sickle-cell disease and osteonecrosis, along with the greater vascularity of the cancellous bone, may make them reasonable candidates for early intervention with core decompression. One report recommended core decompression for relief of chronic hip pain in a series of 13 hips in 10 young patients with a mean age of 15 years (range 9 to 21 years) and a mean follow-up time of 3.7 years (25). Ten of 11 hips without collapse had improvement in pain, and only 3 had radiographic progression. In another report, core decompression was performed in three patients with early stage II osteonecrosis without collapse, but no patient had relief of pain and all required arthroplasty within 1 year (26). A prospective case-control study from 1994 to 2008 concluded that core decompression was an effective way to treat avascular necrosis of the femoral head in patients with sickle-cell disease (27). Their data show pain relief in 39 out of 42 hips. Ten of the 42 hips originally treated with core decompression worsened to the point of requiring total arthroplasty after 7.4 (±2.7) years, compared to the nonoperated controls which required total arthroplasty in 9 out of 23 hips after only 2.6 (±2.4) years. However, a different study reported no difference after a
mean follow-up of 3 years between a group receiving physical therapy and core decompression and a control group that received physical therapy only (28). Longer-term follow-up studies will be required to determine if core decompression significantly alters the natural history of the osteonecrosis associated with sickle-cell disease, or whether physical therapy alone is sufficient.
mean follow-up of 3 years between a group receiving physical therapy and core decompression and a control group that received physical therapy only (28). Longer-term follow-up studies will be required to determine if core decompression significantly alters the natural history of the osteonecrosis associated with sickle-cell disease, or whether physical therapy alone is sufficient.
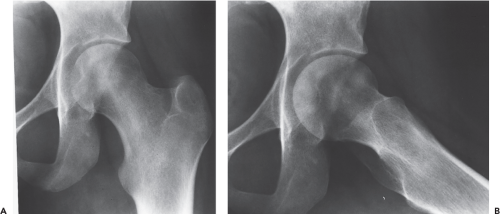 Figure 36.2. The anteroposterior (AP) (A) and lateral (B) radiographs of a 22-year-old woman with sickle-cell anemia and left hip pain show stage II osteonecrosis. The diagnosis was confirmed by MRI scan and histologic examination. |
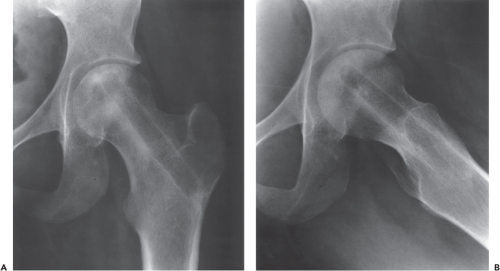 Figure 36.3. The AP (A) and lateral (B) radiographs 3 years after core decompression. Patient has no pain in the hip and there has been no radiographic progression. |
Cementation
An alternative surgical treatment for hips with stage III osteonecrosis has been described in a study from France (19). In 16 hips in 10 patients with sickle-cell hemoglobinopathies, the necrotic bone and articular cartilage overlying it were elevated and then injected with low-viscosity acrylic cement to restore
the sphericity of the femoral head. At a follow-up time of 3 to 7 years (with a mean 5 years), two hips required arthroplasty, one at 1 year and the other at 2 years, but 14 hips were improved. However, some of these patients have pain with walking, or after prolonged activity and limited abduction, with worsening radiographs, indicating that progressive arthritis is present. Further follow-ups from this group and other centers are required before this technique can be recommended.
the sphericity of the femoral head. At a follow-up time of 3 to 7 years (with a mean 5 years), two hips required arthroplasty, one at 1 year and the other at 2 years, but 14 hips were improved. However, some of these patients have pain with walking, or after prolonged activity and limited abduction, with worsening radiographs, indicating that progressive arthritis is present. Further follow-ups from this group and other centers are required before this technique can be recommended.
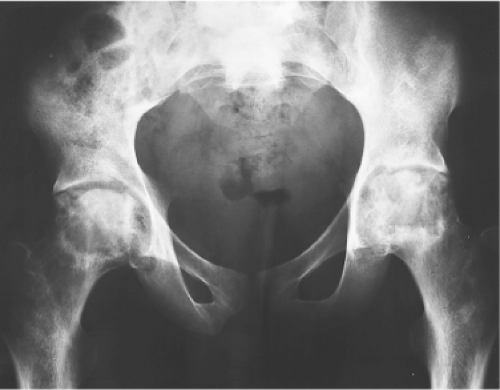 Figure 36.4. Preoperative AP radiograph of the pelvis of a 21-year-old man with sickle-cell disease and endstage osteonecrosis of both femoral heads. Bilateral uncemented total hip arthroplasties were performed for severe pain and disability. |
Prosthetic Hip Resurfacing
Resurfacing of only the osteonecrotic femoral head before acetabular cartilage degeneration has occurred seems an attractive and conservative procedure. Although hemiresurfacing has been successful in short- and medium-term follow-up for osteonecrosis associated with other disorders, it has not been successful in sickle-cell disease. One study reported failure of all four hemiresurfacing procedures in sickle-cell patients (29). One case report described severe protrusio postoperatively, at only 2 years, following hemiresurfacing in a sickle-cell patient (30).
Total Hip Arthroplasty
There is a high prevalence of postoperative complications and a very high rate of failure of all types of hip arthroplasties in patients with sickle-cell hemoglobinopathies. The data from 10 published studies (26,31,32,33,34,35,36,37,38,39) is summarized in Table 36.1. The prevalence of postoperative complications in these studies ranges from 7.7% to 100%, with excessive blood loss, sickle-cell crises, wound drainage, and hematoma frequently reported. Hospitalization is prolonged in most cases. One report described difficulty in preparing the femur, which had sclerotic bone and obliteration of the medullary canal and resulted in the intraoperative complication of three perforations and four femoral fractures (26). The rate of infection is very high: 16% to 20% in several studies, probably due to the high incidence of bacteremia. One study had much lower rates of infection (3%) using prophylatic antibiotics administered intravenously for 3 days, or 30 days if cultures came back positive, as well as antibiotics mixed in with cement for a long release over several years (39). There is a high rate of failure caused by loosening of all types of hip arthroplasties, ranging from 38% at 5 years to a 50% rate of revision at 10 years in some studies. In one series of 13 hips with cemented components, nearly all failed, at a mean follow-up time of 43 months (26). The results with uncemented components were much better, but the follow-up time was only 2 years. In the previously discussed multicenter study of osteonecrosis, 27 patients had a hip arthroplasty, and the probability of survival was only 70% at 4.5 years (16). In one study of 16 primary and revision arthroplasties in these patients, there were no early or late infections at a mean follow-up time of 6 years (36). Two young active men had severe polyethylene liner wear with osteolysis requiring reoperation at 7 and 10 years, respectively (Figs. 36.4 and 36.5). Overall, the result was excellent in nine hips, good in four, and poor in two, with five hips (33%) requiring reoperation during the follow-up period. One study reported the results of 36 bilateral, one-stage bipolar arthroplasties using small uncemented femoral components. At a mean follow-up time of 5.7 years, there was no femoral component loosening, but two complications (protrusio, instability) related to the bipolar shell (37). At the present time, cementless components are strongly recommended for all primary and revision THAs in patients with sickle-cell hemoglobinopathies.
Hemophilia
Etiology
Clotting disorders result from abnormalities in the vascular, platelet, or plasma phases of the clotting process. This is complicated by overlap between these three phases. Besides vessel constriction, the vascular phase involves the exposure of tissue elements that promote platelet activation and activate both the extrinsic and intrinsic plasma phases of the clotting process. The majority of musculoskeletal pathology is caused by X-linked deficiency of factor VIII (classic hemophilia or hemophilia A) or factor IX (Christmas factor or hemophilia B). Repeated hemarthroses lead to synovial hypertrophy, chronic synovitis, cartilage degradation, and eventual arthropathy. The other 11 known plasma coagulation factors may be deficient or absent, but they rarely cause joint hemorrhage without associated trauma (40). Von Willebrand factor, a protein produced by endothelial cells and megakaryocytes, promotes platelet adhesion and serves as a plasma carrier for factor VIII. Factor VIII–related Von Willebrand factor deficiency accounts for less than 1% of hip arthropathy. In a review of 270 hemophiliac patients with orthopedic problems admitted at one center, 236 patients had factor VIII deficiency, 32 patients had factor IX deficiency, and only 2 patients had Von Willebrand’s disease (1 of whom required a total hip replacement [THR]) (41).
Radiographic Findings
One study reviewed the pelvic radiographs of 34 patients (64 hips) from a population of 175 hemophiliac patients
complaining of hip pain (42). Of the hips with an open proximal femoral epiphysis, 80% had a valgus deformity, but none had osteoarthritic changes. Of the skeletally mature hips, 15% had degenerative changes, including protrusio acetabuli in eight hips.
complaining of hip pain (42). Of the hips with an open proximal femoral epiphysis, 80% had a valgus deformity, but none had osteoarthritic changes. Of the skeletally mature hips, 15% had degenerative changes, including protrusio acetabuli in eight hips.
Table 36.1 Results of Total Hip Arthroplasty in Sickle-cell Anemia | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Nonsurgical Management
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


