Soft-Tissue Sarcomas
Murali M. Chintagumpala
The soft-tissue sarcomas form a diverse group of malignant neoplasms that arise from embryonal mesenchyma. As a group, these tumors are rare in children, and most of the information known about these diseases is derived from treating adults. The exception is rhabdomyosarcoma (RMS), a tumor of embryonal mesenchyma that gives rise to striated skeletal muscle. This malignancy is the most common soft-tissue sarcoma of children, and it accounts for 5% to 15% of all malignant solid tumors in patients younger than 15 years. Since the early 1980s, successful treatment regimens have been developed, especially for localized disease, using a combination of surgery, irradiation, and chemotherapy.
RHABDOMYOSARCOMA
The first recorded description of RMS was by Weber in 1854, and the histology of these muscle tumors was described by
Rakov in 1937. Series of patients, mostly adults, were presented by Stout in 1946 and by Pack and Eberhart in 1952. The histology of these tumors was pleomorphic, a type we now know is rare in children. In 1950, Stobbe and Dargeon described the embryonal form of RMS, the most common histologic variety in pediatric patients. In 1958, Horn and Enterline described the four currently recognized histologic subtypes of this malignancy: pleomorphic, embryonal, alveolar, and botryoid.
Rakov in 1937. Series of patients, mostly adults, were presented by Stout in 1946 and by Pack and Eberhart in 1952. The histology of these tumors was pleomorphic, a type we now know is rare in children. In 1950, Stobbe and Dargeon described the embryonal form of RMS, the most common histologic variety in pediatric patients. In 1958, Horn and Enterline described the four currently recognized histologic subtypes of this malignancy: pleomorphic, embryonal, alveolar, and botryoid.
Surgical removal of the primary tumor was the original therapy, and this resulted in some long-term survival. It was found, however, that the malignancy recurred frequently and early in the course of the disease. Survival varied by the site of the primary disease: Head and neck, excluding orbit, had survival rates of 7% to 14%; orbit, 21% to 48%; trunk or extremity, 22%; bladder, 73%; and vagina, 40%. In retrospect, it was evident that, in most cases, the metastases had occurred before the diagnosis could be made.
In 1950, Stobbe and Dargeon reported that at least some RMSs were radiosensitive. In 1959, D’Angio et al. observed a synergistic effect using radiation and dactinomycin. Edland (in 1965) and Sagerman (in 1972) showed that a fractionated total dose of 6,000 cGy could locally control this tumor.
During the same time, reports surfaced that chemotherapeutic agents used singly were successful in producing complete or partial responses in some patients, but the duration of improvement was short. Combined treatment regimens were increasingly more successful. Pinkel and Pickren suggested a coordinated approach to the treatment of RMS using surgery, irradiation, and chemotherapy. The utility of this approach has been confirmed repeatedly.
Because RMS is such a rare disease, the three pediatric groups studying cancer in children pooled their patients and resources to form the Intergroup Rhabdomyosarcoma Study (IRS). The results from the IRS studies have greatly advanced our knowledge of and success in dealing with this disease.
Epidemiology
RMS is the most common of the soft-tissue sarcomas in children, accounting for 4% to 8% of all malignant diseases in children younger than 15 years. RMS is the third most common neoplasm among the extracranial solid tumors of childhood after neuroblastoma and Wilms tumor. The annual incidence is between 4 and 7 cases per 1 million children, with approximately 250 new cases diagnosed in the United States each year. This tumor is 1.4 times more common in boys than in girls. The incidence of RMS appears to be lower in most of Asia than in the white populations of the Western industrialized countries. Relatives of children with RMS have a high frequency of carcinoma of the breast and of brain tumors. RMS has been associated with certain familial syndromes, such as neurofibromatosis and the Li-Fraumeni syndrome, which is associated with germline mutations of the p53 gene. The use of marijuana, cocaine, or any other recreational drug by one or both parents has been shown to be associated with an increased risk of RMS in the child.
Clinical Manifestations and Complications
RMS can occur anywhere in the body. The percentage of cases presenting at each anatomic location is depicted in Table 309.1. The head and neck (including the orbit) are the most common sites of primary occurrence, with 38% of the cases presenting in this region. The orbit accounts for 10% of the total presentations. The genitourinary tract is next in order of frequency, followed by the extremities, trunk, retroperitoneum, and other sites.
TABLE 309.1. PRIMARY SITES OF RHABDOMYOSARCOMA | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Approximately two-thirds of the tumors occur in children 6 years or younger, with a peak incidence between the ages of 2 and 5 years. The signs and symptoms relate to the primary site of the tumor or the metastases. Usually a painless, enlarging mass is noticed.
Tumors in the orbit can produce proptosis, chemosis, and ocular paralysis. These tumors can begin as a mass in the conjunctiva or eyelid. Tumors in the nasopharynx can cause a nasal voice, dysphagia, airway obstruction, epistaxis, or pain. Tumors in the paranasal sinuses cause swelling, pain, discharge, sinusitis, obstruction, or epistaxis. Laryngeal tumors cause hoarseness. Tumors in the middle ear are associated with a polypoid tumor in the external auditory canal that can causes pain, chronic otitis media, and a facial nerve palsy. RMS may present as a painless facial or parotid mass. Patients with neck masses may present with hoarseness or dysphagia. Parameningeal tumors may extend into the central nervous system (CNS), resulting in meningeal symptoms, cranial nerve palsies, or respiratory paralysis.
Tumors arising from the trunk, extremities, or paratesticular region usually occur as painless masses that are noticed by the child or parents. Tumors in the retroperitoneum usually are asymptomatic or are found as large masses that may cause gastrointestinal or urinary tract symptoms. Bladder and prostate tumors usually produce urinary tract symptoms. Tumors from the perineum may involve the bowel or bladder. Botryoid tumors appear as grapelike clusters of clear tissue protruding from the uterus or cervix.
The tumor characteristically grows with indistinct margins along fascial planes and infiltrates into surrounding tissues. Metastases spread hematogenously and by lymphatics to the lung, bone, bone marrow, lymph nodes, CNS, heart, and breast.
Diagnosis
Open biopsy of the tumor is the definitive diagnostic procedure for an unexplained mass. Certain tests are performed before the surgical procedure to assess the extent of the disease for staging and therapeutic purposes.
Preoperative assessment should include a complete blood count, urinalysis, measurement of electrolytes (including calcium and phosphorus), liver and renal function tests, and a uric acid determination. Computed tomography (CT) or magnetic resonance imaging (MRI) of the primary tumor should be performed to delineate the involvement of adjacent structures and to aid in the surgical management of the patient. A CT scan of the chest, bone marrow examination, bone scan or skeletal survey, and liver scan should be performed to look for metastases. Patients with cranial parameningeal tumors also should
have a CT scan and/or MRI of the head and an examination of the cerebrospinal fluid (CSF) to look for evidence of meningeal seeding with CSF pleocytosis, elevation in protein, and reduction of glucose. A CT scan may be used to assess retroperitoneal lymph node involvement in patients with lower extremity and genitourinary tumors. Figure 309.1 shows a solid tumor in the pelvis that was demonstrated to be RMS after surgical biopsy.
have a CT scan and/or MRI of the head and an examination of the cerebrospinal fluid (CSF) to look for evidence of meningeal seeding with CSF pleocytosis, elevation in protein, and reduction of glucose. A CT scan may be used to assess retroperitoneal lymph node involvement in patients with lower extremity and genitourinary tumors. Figure 309.1 shows a solid tumor in the pelvis that was demonstrated to be RMS after surgical biopsy.
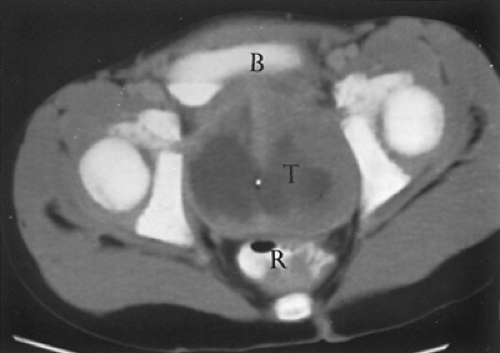 FIGURE 309.1. In the computed tomographic scan of a patient with a pelvic mass that was shown to be rhabdomyosarcoma after a surgical biopsy, the large pelvic tumor (T) is possibly associated with the prostate, displacing the rectum (R) posteriorly and the bladder (B) anteriorly, and extending to the side walls bilaterally. Notice the Foley catheter in the center of the tumor and the central area of necrosis depicted by the darker region of the mass. |
Histology
Histologically proven RMS on gross examination does not differ from other malignant soft tissue tumors and, with the exception of the grapelike clusters of sarcoma botryoides, the tumors do not differ from each other. The tumors are firm, nodular, and grossly well circumscribed but not encapsulated, and they aggressively invade adjacent tissues.
Four histologic variations have been described. The most common form is the embryonal type, which consists of spindle-shaped myoblasts and small round cells. This type accounts for 57% of RMSs and 75% of the tumors arising from the head and neck and genitourinary tract. Patients with this histologic variant have a relatively favorable prognosis.
The alveolar type is the second most common type of tumor, accounting for approximately 20% to 30% of the cases. The alveolar type is seen more commonly in children older than 6 years and occurs most often in the trunk, extremities, and perianal region. These tumor cells grow in cords that often have cleftlike spaces resembling alveoli. Patients with this tumor have a poorer prognosis than those with the embryonal type.
The botryoid type is most often seen in the genitourinary tract. This tumor accounts for approximately 6% of cases and is seen more commonly in children younger than 6 years. A deep, compact zone of spindle-shaped cells resembling myoblasts is found under a layer of myxoid stroma with a layer of small round cells at the periphery. This tumor is associated with a prognosis similar to the embryonal type.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


