Skeletal Dysplasias
Patricia G. Wheeler
David D. Weaver
Skeletal dysplasias are conditions presenting primary problems in growth resulting from defective formation of bone or cartilage. This category of diseases includes a heterogenous group of disorders with a wide variety of clinical and radiographic manifestations. Much of the variation stems from the different combinations of involved bones and the ways in which these bones are affected in their shape, length, and density. Most skeletal dysplasias are genetically determined, can be inherited, and result in disproportionately short stature. Because of the last problem, the term dwarfism frequently is applied to these disorders. Other names include chondrodysplasias, osteochondrodysplasias, and bone dysplasias.
More than 200 recognized types of skeletal dysplasias exist. The incidence of any single dysplasia is relatively low. Collectively, however, they are relatively frequent (1 in 3,000 to 5,000 births). Because of this frequency and the diversity of these conditions, it is essential that the practitioner be able to recognize a fetus, newborn, or child with a skeletal dysplasia so that the diagnosis can be established, the prognosis determined, and a treatment plan developed. Genetic counseling should be
available to the individual, parents and family in regards to recurrence risks.
available to the individual, parents and family in regards to recurrence risks.
This chapter provides the primary-care physician with a basic understanding of skeletal dysplasias and presents the major clinical and radiographic features and complications of the more commonly encountered disorders. When encountering one of these disorders, the responsibilities of the primary-care physician are to treat any immediate medical problems, become familiar with the particular skeletal dysplasia present in the patient, and obtain appropriate and timely consultation. In all cases in which the patient survives the neonatal period, a long-term treatment program should be established.
EPIDEMIOLOGY
Most skeletal dysplasias are uncommon, and often their exact incidence is unknown. Osteogenesis imperfecta (OI or brittle bone disease), type I, which is one of the milder forms of the disorder, has an estimated frequency of 1 in 20,000 births. OI type II, the more severe and normally lethal form, is less common and affects approximately 1 in 50,000. Thanatophoric dysplasia, the most frequently encountered lethal skeletal dysplasia, is seen in approximately 1 in 30,000 births. Classic or heterozygous achondroplasia, the most common nonlethal skeletal dysplasia, occurs with a frequency of 1 in 25,000.
On the other hand, the incidence of some skeletal dysplasias in certain ethnic groups may be much higher. For instance, the McKusick type of metaphyseal chondrodysplasia (cartilage-hair hypoplasia) is rare in the general population, but is found in approximately 1 in 500 live births among the Amish population of North America. The higher incidence of certain bone dysplasias in some ethnic groups normally is the result of inbreeding or other factors that have increased the gene frequency in that group.
PATHOGENESIS
Development and Anatomy of Bone
Bone is formed from either mesodermal or neural crest cells. These cells form mesenchyme, which in turn forms embryonic connective tissue. For cartilaginous bone, cartilage is formed within the embryonic connective tissue, which then changes to bone. For membranous bone, the bone is formed directly within the connective tissue. The cartilage in cartilaginous bone is formed by chondroblasts, whereas the mineral portion of the bone is derived from osteoblasts in both bone types. The skull (except for its base), maxilla, mandible, squamous portion of the temporalis, nasal bones, and clavicles are membranous bone; all the rest of the skeleton is cartilaginous bone.
The anatomy of long bones is depicted in Figure 433.1. When long bones are growing, the growing end is known as the epiphysis. Below the epiphyseal center is the metaphysis. Longitudinal growth actually occurs at the junction between the epiphysis and the metaphysis, an area called the physis or growth plate. During puberty, the growth plate is obliterated, the epiphysis and metaphysis fuse, and lengthening of the bone ceases. The shaft of the bone is called the diaphysis. Molding of the diaphysis takes place as the bone becomes longer. The diaphysis also thickens with age and in response to increased stress exerted on the bone.
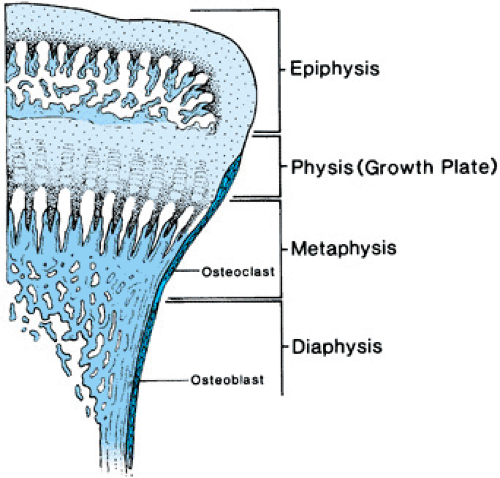 FIGURE 433.1. Anatomy of normal prepubertal tubular bone. Note that the bone is divided into four segments, each of which may be involved in skeletal dysplasias either separately or in combination. |
Nomenclature and Classification of Skeletal Dysplasias
Skeletal dysplasias in general are named after the anatomic parts of the bones that are affected, after the appearance of the bone, or after the individual(s) who originally described the condition (Fig. 433.2). In some cases, a combination of these methods is used.
An example of the anatomic naming of bone dysplasias is multiple epiphyseal dysplasia (Fig. 433.3). In this condition,
numerous epiphyses (see Fig. 433.2B) are involved, whereas the spine is affected only minimally. As a result of the involvement of multiple epiphyses, the individual usually has short stature and, with age, may develop significant arthritis in the involved joints. Another example of anatomic nomenclature is spondyloepiphyseal dysplasia congenita, in which many of the epiphyses, including those of the spine (spondylo-), are abnormal. The clinical picture is one of small, irregular epiphyses, flattened vertebral bodies (platyspondyly; see Fig. 433.2E, F) and, in some cases, kyphoscoliosis.
numerous epiphyses (see Fig. 433.2B) are involved, whereas the spine is affected only minimally. As a result of the involvement of multiple epiphyses, the individual usually has short stature and, with age, may develop significant arthritis in the involved joints. Another example of anatomic nomenclature is spondyloepiphyseal dysplasia congenita, in which many of the epiphyses, including those of the spine (spondylo-), are abnormal. The clinical picture is one of small, irregular epiphyses, flattened vertebral bodies (platyspondyly; see Fig. 433.2E, F) and, in some cases, kyphoscoliosis.
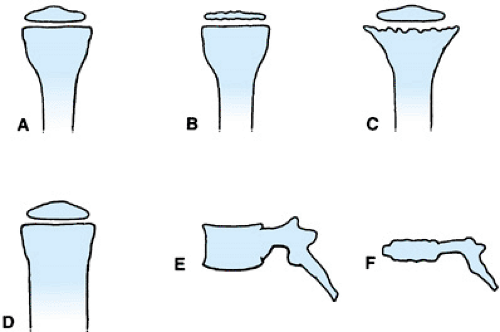 FIGURE 433.2. Diagrammatic representation of various types of bone dysplasias. A: Normal shape of growing long bones. B: Epiphyseal involvement such as in multiple epiphyseal dysplasia; note flattened and irregular epiphysis. C: Metaphyseal involvement that might be seen in achondroplasia; note widening and irregular surface of metaphysis. D: Diaphyseal involvement as seen in craniodiaphysial dysplasia. E: Normal lateral view of vertebra. F: Vertebral involvement that can be found in spondyloepiphyseal dysplasia congenita; note vertebral body flattening and surface irregularities. (Modified with permission from Rimoin DL, Lachman RS. The chondrodysplasias. In: Emery AEH, Rimoin DL, eds. Principles and practice of medical genetics. New York: Churchill Livingstone, 1990:895.) |
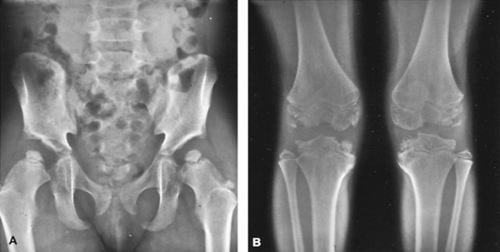 FIGURE 433.3. Radiographs of a patient with multiple epiphyseal dysplasia. A: Pelvis and hips at age 8 years; note small irregular proximal femoral epiphyses and poorly developed acetabular roof. B: Knees of patient at age 10 years; note irregular and mottled epiphyses of the long bones. |
Other bone dysplasias are named after the appearance of the bone. An example is campomelic (or camptomelic) dysplasia. In this disorder, a characteristic bowing of the long bones of the lower extremities occurs. The term camptomelic is coined from the Greek campo, which means bent or curved. In OI, the term imperfecta refers to poorly mineralized bone. In a newly described disorder, schneckenbecken dysplasia, the pelvis is shaped like a snail shell; schneckenbecken is German for “snail-shaped.”
The third mechanism for naming bone dysplasias involves the use of eponyms. For example, Kniest dysplasia is a rare, autosomal dominant disorder characterized by short stature and dumbbell-shaped femurs with extremely broad and shortened femoral necks. Kniest first reported the condition in 1952.
Frequently, eponyms are used in combination with one of the first two methods of naming skeletal dysplasias, most often to delineate the particular subtype of dysplasia. For instance, two types of achondrogenesis exist, one known as Parenti-Fraccaro achondrogenesis (type I) and the other as Langer-Saldino achondrogenesis (type II; Fig. 433.4). These two subtypes are not based on differences in the clinical appearance of the affected children (both types have nearly identical clinical phenotypes), but on radiographic variations. Type I has been subdivided further into type IA, with rib fractures and spike-shaped femurs; and type IB, without the rib fractures but with a pelvis that appears to be turned upside down. Type IA also has been denoted as the Houston-Harris type and type IB as the Fraccaro type.
Various systems for classifying the skeletal dysplasias have been devised. One of the earliest of these was based on the relative shortening of the limb or spine: For example, was the short stature of the child caused by primary shortening of the extremities, with relatively normal spinal length, or was the spine relatively short compared with the length of the arms and legs?
Achondroplasia is one example of a short-limbed skeletal dysplasia. In this condition, the sitting height—that is—the length of the trunk and head of an individual when she is sitting, is nearly normal, but the standing height is far less than normal. This reflects the very short legs in this condition. On the other hand, patients with Morquio syndrome (mucopolysaccharidosis, type IV) have the opposite characteristics. They have short trunk and, in relationship to the spinal length, long extremities. The shortening of the spine in Morquio syndrome is primarily a result of platyspondyly.
Another classification system is based on the age at which the first manifestation of the skeletal dysplasia appears. Some disorders can be detected prenatally or at birth (congenita), whereas others do not have clinical or radiographic manifestations until months or years after birth (tarda). Yet another system is based on the pattern of bone involvement and the types of skeletal changes seen in a particular disorder. The International Nosology and Classification of Constitutional Disorders of the Bone (Table 433.1) use this approach.
Yet another approach has been put forth by Kornak and Mundlos. Utilizing recent advances in molecular genetics, these authors have presented a classification based on a combination of molecular pathology and embryology. Bones develop first by the formation of a pattern: the determination of the location, size and shape of individual bones. Next, mesenchymal cells differentiate and condense into cartilage. Third, growth occurs in the cartilaginous growth plates at the ends of long bones. Finally, remodeling and homeostasis occurs. These authors have classified bone disorders according to how these disorders affect these four phases of bone development and maintenance. Bone disorders also may be classified as lethal and nonlethal.
Dysostoses
Dysostoses are conditions predominately affecting individual bones, either singly or a few in combination. A specific bone may be absent, hypoplastic, or malformed. Further dysostoses are distinguished from more generalized skeletal dysplasias
by being formed during embryogenesis (the third through the eighth week post conception), whereas skeletal dysplasias generally develop throughout intrauterine development and childhood.
by being formed during embryogenesis (the third through the eighth week post conception), whereas skeletal dysplasias generally develop throughout intrauterine development and childhood.
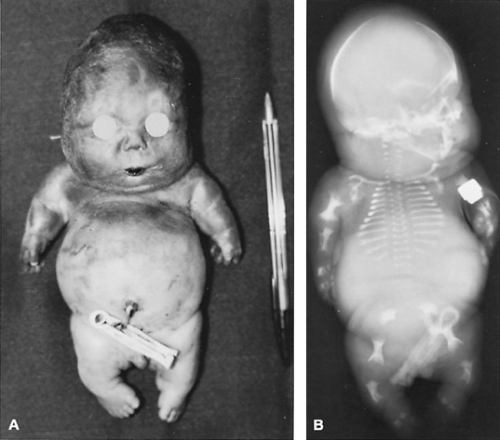 FIGURE 433.4. Achondrogenesis, type II in a term newborn. A: Note severe growth deficiency (standard ballpoint pen alongside for comparison), small chest and extremities, and protruding abdomen. This was a postmortem picture. B: Radiograph of the same patient; note the very short, irregularly shaped bones of the extremities, short and horizontal ribs, and poorly calcified vertebral bodies. |
Delineation of Skeletal Dysplasias
The discovery of new skeletal dysplasias has been an ongoing process since the 1940s. As noted already, a bewildering number of different disorders are recognized, making diagnosis problematic for the average clinician. The recognition of a specific disorder further is complicated by the similar appearances of many of these conditions. Furthermore, a spectrum of severity may exist within a single entity, or two conditions may have overlapping features, making arrival at a correct diagnosis exasperating even for the experts. A further characteristic of many skeletal dysplasias is that these conditions often become more severe with time. The condition may manifest prenatally, and may be severe, if not lethal, at birth. If the disorder is not lethal or not expressed at birth, it invariably worsens with age. If a child is seen early in the course of the condition, when the features often are not expressed fully, one may find it difficult or impossible to make the diagnosis.
During the 1940s, most individuals with disproportionately short stature were classified as having achondroplasia if the extremities were relatively short, or as having Morquio disease if the trunk was shortened disproportionately. Since then, numerous different skeletal dysplasias with similar appearances have been delineated from each of these disorders. This demarcation has been accomplished by the careful study of clinical, radiographic, and pathologic features, and noting the inheritance patterns of each. A newer trend in the grouping of skeletal dysplasias involves categorizing each skeletal dysplasia based on the specific protein, enzyme, or gene defect involved, if known.
The biochemical and/or genetic delineation of numerous skeletal dysplasias has made it clear that many previously described and well-characterized conditions are, in actuality, the result of mutations at different locations within the same gene. The opposite also is true; individuals with the same apparent clinical condition may be biochemically and molecularly distinct. This heterogeneity in part accounts for the difference in severity of the “same” clinical condition.
Molecular Genetics and Biochemistry of Skeletal Dysplasias
During the last two decades, much has been learned about the molecular genetics and biochemistry of many skeletal dysplasias. Biochemical errors have been found in the structural proteins of cartilage (specifically different types of collagen), enzymes of cartilage metabolism (lysosomal enzymes), and regulators of cartilage growth (fibroblast growth factor receptors). As other genes and biochemical pathways in bone formation and function are found, one would expect to find additional errors in the bone matrix, osteoid (the mineral portion of the bone), and other components of cartilage. In addition, problems may be found in the function of the osteoblasts (those cells that form bone) or the osteoclasts (those cells that degrade and reshape bone).
One of the first skeletal dysplasias to be understood on the molecular and biochemical levels was OI. This disorder is characterized by bone fragility resulting in fractures. OI is divided into four main types based on clinical and radiographic findings. Type II is normally a perinatal or neonatal lethal, with multiple fractures occurring in utero, and the affected newborn typically having marked respiratory distress because of a small and excessively flexible chest and short and curved long bones. OI type III also presents at birth with multiple fractures, but respiratory complications are not as severe as in type II. Individuals with OI type III typically have multiple broken bones occurring throughout their life, progressive bone deformation, and marked short stature. OI types I and IV are less severe, with fewer fractures and most fractures occurring postnatally;
short stature and teeth abnormalities also may be present. All four types of OI are produced by a defect in either the procollagen pro-alpha1 gene (COL1A1) or the procollagen pro-alpha2 gene (COL1A2). These genes normally produce the polypeptides pro-alpha1 and pro-alpha2, respectively. Under normal circumstances, two pro-alpha1 chains combine with one pro-alpha2 procollagen in a helical structure to form type I procollagen (Fig. 433.5). The end portions of the type I procollagen molecules then are cleaved to form mature procollagen molecules that are then secreted into the extracellular matrix and incorporated into the definitive type I collagen fibril. Deletions of one or more amino acids, or the substitutions of amino acids in the pro-alpha1 or pro-alpha2 chains, have been found in most patients with OI.
short stature and teeth abnormalities also may be present. All four types of OI are produced by a defect in either the procollagen pro-alpha1 gene (COL1A1) or the procollagen pro-alpha2 gene (COL1A2). These genes normally produce the polypeptides pro-alpha1 and pro-alpha2, respectively. Under normal circumstances, two pro-alpha1 chains combine with one pro-alpha2 procollagen in a helical structure to form type I procollagen (Fig. 433.5). The end portions of the type I procollagen molecules then are cleaved to form mature procollagen molecules that are then secreted into the extracellular matrix and incorporated into the definitive type I collagen fibril. Deletions of one or more amino acids, or the substitutions of amino acids in the pro-alpha1 or pro-alpha2 chains, have been found in most patients with OI.
TABLE 433.1. SKELETAL DYSPLASIAS | ||
|---|---|---|
|
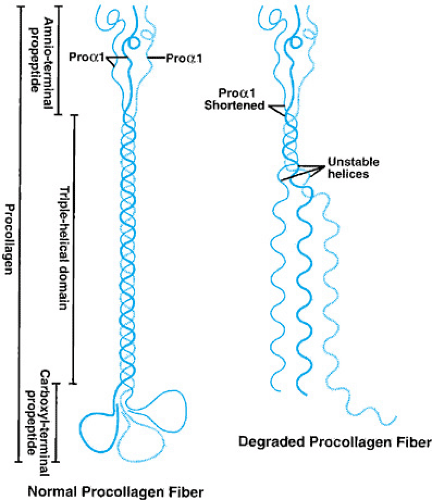 FIGURE 433.5. Normal type I procollagen molecule on the left showing normal helical formation. The figure on the right depicts the lack of coiling of the polypeptides when a change in or deletion of one or more amino acids occurs in both pro-alpha1 chains, leading to degradation of the molecule. One pro-alpha1 chain in each figure has been thickened for clarity. |
When these defects occur in the pro-alpha1 or pro-alpha2 chains, the formation of the normal helical structure of the type I procollagen molecule frequently is disrupted (see Fig. 433.5). If the disruption is significant enough, the abnormally formed procollagen is degraded within the cell, a process called protein suicide. The end result of protein suicide is a deficiency of type I collagen, but the type I collagen that is formed is normal. When the disruption of the helical formation is less severe, intracellular degradation of the procollagen fiber may not occur, and the abnormal procollagen is secreted from the cell and incorporated into the collagen fiber, thus making the collagen abnormal. Studies have shown that individuals who have more severe mutations (for example, premature stop codons or a substitution of a glycine) that prevent a pro-alpha1 or a pro-alpha2 chain from being formed typically have a milder form of OI (OI type I or IV), whereas those who have less disruptive mutations (single base-pair changes leading to substitution of a nonglycine amino acid) that allow the abnormal protein to be incorporated into the procollagen strand tend to have a more severe form of OI (type II or III). The clinical phenotype appears to be milder if only 50% of the normal collagen protein is present, as opposed to having a normal amount of abnormally formed collagen. In reality, OI is a continuous spectrum of severity with type II being at one end and normally an early lethal condition, and types I and IV being at the other, with some individuals having few problems and a normal lifespan. This spectrum in part is due to the fact that over 200 mutations have been reported in COL1A1 and COL1A2 genes. However, the severity in any one family is frequently often similar, because affected individuals normally possess the identical mutations (the exception being type IV, in which striking intrafamilial variability may be present).
Until recently, isolated cases of possible OI were evaluated by looking for the abnormal or reduced production of procollagen in tissue samples, usually through skin biopsy from affected individuals. This evaluation detected approximately 85% of individuals with OI. However, direct gene testing of COL1A1 or COL1A2 now is available. Testing for these genes can be done on a blood sample and involves sequencing of both the COL1A1 and COL1A2 genes. This testing finds a mutation in 95% of individuals with clinical OI.
Another bone dysplasia that has been studied in detail is achondroplasia, the most common nonlethal skeletal dysplasia causing short stature. The genetic defect in achondroplasia has been isolated to a specific mutation in the fibroblast growth factor receptor 3 (FGFR3) gene (Figs. 433.6 and 433.7). In more than 99% of analyzed achondroplastic cases, a glycine-to-arginine substitution has been found at amino acid position 380 of the receptor. On the other hand, mutations in other areas of the FGFR3 gene result in a lethal bone dysplasia, thanatophoric dysplasia, and a milder skeletal dysplasia,
hypochondroplasia. The location of these and other mutations in the FGFR3 gene are depicted in Fig. 433.7.
hypochondroplasia. The location of these and other mutations in the FGFR3 gene are depicted in Fig. 433.7.
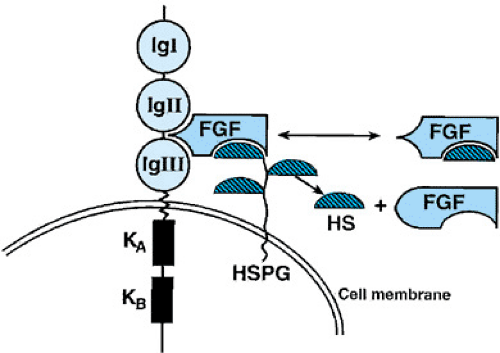 FIGURE 433.6. Model of fibroblast growth factor receptor 3 (FGFR3). The ligand, fibroblast grown factor (FGF), first binds to heparan sulfate proteoglycan (HSPG) (hashed half ovoids attached to the cell membrane), or heparan sulfate (HS). The complex then forms a trimolecular complex with FGFR3, resulting in structural changes in the tyrosine kinase parts of the molecules (KA and KB) and sequences genetic regulation of the cell. The IgI, IgII, and IgIII regions represent immunoglobin-like loops involved in the attachment of the FGF-HS/FGF-HSPG complex. The 23 recognized FGFs are structurally related polypeptides that play various roles in bone embryogenesis, growth, and homeostasis by inducing differentiation and mitogenesis, and stimulating chemotaxes, cell survival, and angiogenesis. Tyrosine kinase domains are involved in intracellular signalling. (Modified with permission from Givol D, Yayon A. Complexity of FGF receptors: genetic basis for structured diversity and functional specificity. FASEB J 1992;6:3362.) |
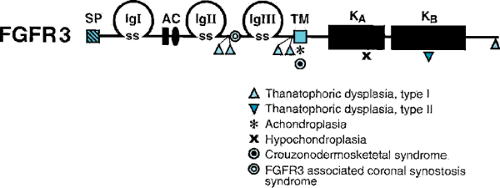 FIGURE 433.7. Location of mutations found in fibroblast growth factor receptor 3 (FGFR3) protein resulting in the indicated bone dysplasias. Achondroplasia is produced at only one location, the transmembrane TM domain, whereas thanatophoric dysplasia, type I results from mutations at five different sites. Abbreviations starting from the left: SP, signal peptide; SS, sulfide bonds keeping the IgI, IgII and IgIII immunoglobin-like loops in their looped form. KA and KB are tyrosine kinase domain involved in intracellular signaling. (Modified with permission from Givol D, Yayon A. Complexity of FGF receptors: genetic basis for structured diversity and functional specificity. FASEB J 1992;6:3362 and Cohen MM Jr. Fibroblast growth factor receptor mutations. In: Cohen MM Jr., MacLean RE, eds. Craniosynostosis: diagnosis, evaluation, and management, 2nd ed. New York: Oxford University Press, 2000:77.) |
One current hypothesis to explain why different mutations in the FGFR3 gene cause different bone dysplasias is based on the mutations causing a range of tyrosine kinase activity (referred to as a gain of function mutation). Tyrosine kinase acts as a negative regulator for endochondral bone growth. The more “turned on” the tyrosine kinase, the greater the reduction in bone length, and thus the greater the severity of the bone dysplasia. Different mutations in the FGFR3 gene lead to a varying degree of up-regulation of the tyrosine kinase and result in the various dysplasias associated with this gene.
Another recent advance in the understanding of normal and abnormal bone growth and the cause of certain bone dysplasias has been the discovery of the SHOX gene. SHOX is the abbreviation for short stature homeobox-containing gene. This gene is located in the pseudoautosomal region of the p-arm of the X and Y chromosomes. The pseudoautosomal regions in these chromosomes contain homologous genes, one gene on the X and one on the Y chromosomes. In females, the genes on the pseudoautosomal regions are not inactivated on the inactivated X-chromosome and, as a result, in both males and females both homologous genes are active. In Turner syndrome, in which only an X chromosome is present or in which the p arm of one X chromosome is missing or partially missing, only half the normal SHOX gene is produced; this in turn leads to the short stature seen in this condition. Furthermore, deletions and point mutations of this gene can lead to idiopathic short stature (ISS) or certain bone dysplasias. Deletions leading to ISS seem to be relatively common, and recently it has been shown that 7% of ISS cases are associated with deletions in the SHOX gene. In individuals with these deletions, the radiographic appearance of the skeleton and growth hormone secretion are normal. Of further interest is that two related skeletal dysplasias also have been found to have mutations or deletions in this gene. The first condition is Leri-Weill dyschondrosteosis, an autosomal dominant disorder, characterized by Madelung deformity of the wrists (bowing of the radius and distal hypoplasia of the dorsally dislocated ulna) and short stature, with most of the shortening in the forearms (mesomelic shortening). In roughly 80% of Leri-Weill dyschondrosteosis cases, a deletion of the gene occurs; in about 20% of cases, a point mutation occurs. A second, rare disorder is mesomelic dysplasia, Langer type, which is the homozygous dominant state of the Leri-Weill dyschondrosteosis; it is caused by mutations in or deletion of both SHOX genes. Why this range in phenotypic expression is associated with the various mutations and deletions of this gene has not been unraveled.
Other biochemical defects also are known to cause skeletal dysplasias. In some forms of hypophosphatasia, a complete or partial deficiency of bone alkaline phosphatase occurs, thus leading to undermineralized osteoid. Vitamin-, mineral-, and hormone-deficient states also can produce skeletal dysplasias. One of the most severe bone dysplasias known (cretinism) is produced by the total absence of thyroxine. The dysplasia occurs because thyroxine appears to be one of the more important hormones involved in normal bone growth, and its deficiency produces a severe delay in osseous maturation.
With the continued application of DNA technologies, the identification of more of the defective genes producing bone dysplasias has been possible. Table 433.2 lists selected genes causing bone dysplasias, their chromosomal locations, and the gene products. In the future, the identification of additional genes will lead to further elucidation of the various biochemical defects, more reliable and specific methods of diagnosing bone dysplasias prenatally and postnatally and, ultimately, methods of treating or preventing these disorders.
DIAGNOSIS
The diagnosis of a skeletal dysplasia should be considered in any patient who has disproportionately short stature or abnormal development of one or more bones. When one encounters such an individual, that individual should be evaluated fully to determine whether a skeletal dysplasia is present and, if so, which one. The history should include a pregnancy history, with particular reference to any fetal evaluation; birth length, weight, and head circumference; postnatal growth and development; and medical problems. With regard to the family history, one should inquire about short stature and orthopedic problems in other family members and determine if consanguinity exists between the parents. While performing the physical examination, one should take care to search for nonosseous abnormalities such as cataracts, cleft palate, and congenital heart disease that would be useful in establishing the diagnosis and be helpful in formulating a treatment plan. Basic measurements should be obtained, including height, weight, head circumference, arm span, and upper segment–to–lower segment ratio. The lower segment length is determined by measuring the distance from the anterior-superior edge of the pubic symphysis to the floor while the patient is standing, or to the bottom of the feet in infants. The upper segment length is obtained by subtracting the lower segment length from the total body length. The upper segment–to–lower segment ratio is obtained by dividing the upper segment length by the
lower segment length. The upper segment–to–lower segment ratio in normal children varies from 1.6 at birth to 0.93 in older teenagers and adults. Many children with bone dysplasias have ratios that are greater than normal for their age as a result of having short lower extremities, whereas children with values less than normal for their age usually have short trunks.
lower segment length. The upper segment–to–lower segment ratio in normal children varies from 1.6 at birth to 0.93 in older teenagers and adults. Many children with bone dysplasias have ratios that are greater than normal for their age as a result of having short lower extremities, whereas children with values less than normal for their age usually have short trunks.
TABLE 433.2. GENES, CHROMOSOMAL LOCI, AND PEPTIDES, PROTEINS, AND ENZYMES INVOLVED IN SKELETAL DYSPLASIAS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


