Category
Mechanism
Treatment
Hemorrhagic
Loss of circulating blood volume
Intravascular volume replacement
Restore O2 carrying capacity
Traumatic
Loss of circulating blood volume with tissue injury and release of DAMPs
Control of bleeding and intravascular volume replacement
Cardiogenic
Cardiac failure
Improve cardiac perfusion, may require vasopressors/inotropes or devices (IABP/LVAD)
Neurogenic
Brain or spinal injury with disruption of the sympathetic regulation of the cardiovascular system
Intravascular volume replacement and vasopressors
Obstructive
Impaired right ventricular diastolic filling or obstruction of right ventricular output
Early diagnosis and resolution of decreased venous return/ventricular filling
Septic
Decrease in systemic vascular resistance with release of PAMPs
Intravascular volume replacement and vasopressors
Pathophysiology of Shock
The pathophysiology of shock is complex and is ultimately driven by tissue hypoperfusion leading to altered cellular metabolism and activation of the immune system. Prolonged ischemia has many detrimental effects at the cellular level (Table 12.2) but eventually results in a shift to anaerobic metabolism and a failure to synthesize adenosine triphosphate (ATP) for maintaining homeostasis [11]. Although the results of ischemia are detrimental, the restoration of blood flow and reoxygenation intensifies the inflammatory response and exacerbates tissue injury [12]. This phenomenon is commonly known as ischemia/reperfusion injury. Ischemia initially results in impaired endothelial cell barrier function and increased vascular permeability, but it is the reperfusion which leads to the activation of cell death pathways, autophagy-associated cell death, and necrosis [13, 14]. These apoptotic pathways are highly regulated by ischemia-induced transcription of genes, such as NF-κ[kappa]B and HIF [15].
Table 12.2
Cellular effects of ischemia
Cellular acidosis |
Altered intracellular ion distribution |
Altered membrane potential |
Cellular swelling |
Cytoskeletal disorganization |
Increased hypoxanthine |
Decreased oxidative phosphorylation and ATP synthesis |
Decreased phosphocreatine |
Decreased glutathione |
Increased nucleotide phosphohydrolysis and adenosine signaling |
Increased leukocyte adhesion molecule expression |
Ischemia/reperfusion injury leading to apoptosis and necrosis is highly immunostimulatory and leads to inflammatory cell infiltration and cytokine production. Release of intracellular products from injured cells such as high-mobility group box 1, heat shock proteins, mitochondrial peptides, heparin sulfate, and RNA has paracrine and endocrine-like effects on distant tissues stimulating inflammatory responses [16]. These molecules that are released have been termed damage-associated molecular patterns (DAMPs), and their effects are physiologically known as danger signaling. DAMPs are recognized by cell surface pattern recognition receptors, such as Toll-like receptors (TLRs), which through intracellular signaling amplify the immune response. Remarkably, similar effects are generated from molecules associated with pathogens, such as bacterial lipopolysaccharide, termed pathogen-associated molecular patterns (PAMPs).
Tissue-resident macrophages or mast cells are the first local cellular responders to ischemia releasing eicosanoids, histamines, and cytokines (IL-1, IL-6, MIP, TNF-α[alpha]) and chemokines (IL-8), which further amplify the immune response (Fig. 12.1) [17]. Macrophage activation subsequently leads to neutrophil priming, chemotaxis, leukocyte-endothelial cell adhesion, and transmigration into the ischemic/injured tissue resulting in further release of TNF-α[alpha] and other cytokines [18]. However, tissue-resident macrophages may take several days to achieve their full inflammatory response, and the complement cascade may play a larger role in the initial inflammatory process [19].
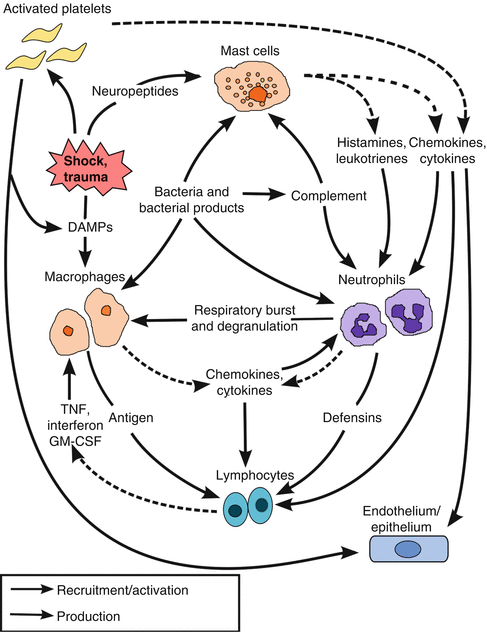
Fig. 12.1
Flow of signaling following trauma and shock resulting in the recruitment and activation of the innate immune system and the amplification of the inflammatory process. DAMPs danger-associated molecular patterns, TNF tumor necrosis factor, GM-CSF granulocyte-macrophage colony-stimulating factor
The complement cascade is activated via several pathways following severe injury, hemorrhagic shock, or infection and produces anaphylatoxins (C3a and C5a), which further recruit and activate macrophages but are also potent attractants and activators of neutrophils. In both injured patients and patients in hemorrhagic shock, the degree of complement activation is proportional to the magnitude of injury or depth of shock [20–22]. Generally, the complement system is known to be activated by either the classical, alternative, or lectin pathways [23]. Activation of the classical pathway requires the formation of antigen-antibody complexes, which binds C1 and initiates the cascade. Following ischemia/reperfusion, cells can express neoepitopes (β[beta]2-glycoprotein, β[beta]-actin, annexin IV, and non-muscle myosin heavy chain type II) or form microparticles, which are bound by natural antibodies and activate complement [24]. The lectin pathway is activated by mannose residues on bacterial surfaces, which bind the mannose-binding lectin (MBL). This complex subsequently activates the MBL-associated serine proteases (MASP-1 and MASP-2), which act similar to the C1 complex. The alternative pathway may be activated by cell surface factors on yeast or bacteria or may be activated spontaneously. Simply, these three pathways converge to a common pathway with the formation of C3 and C5 convertases, which cleave C3 to form C3a and C3b and C5 to form C5a and C5b (Fig. 12.2) [25]. The binding of C3a and C5a to their corresponding receptors (C3aR, C5aR, and C5L2) on both myeloid and nonmyeloid cells incites proinflammatory signaling which acts as a chemoattractant for neutrophils, activates an oxidative burst and lysosomal enzyme release, stimulates mast cell and basophil degranulation, induces expression of adhesion molecules on endothelial cells, promotes smooth muscle cell contraction, and enhances the acute phase response of the liver [26–28]. C3b acts as an opsonin for both prokaryotic and eukaryotic cells, and C5b initiates the assembly of the membrane attack complex (MAC), composed of C5b–C9, which promotes cellular lysis.
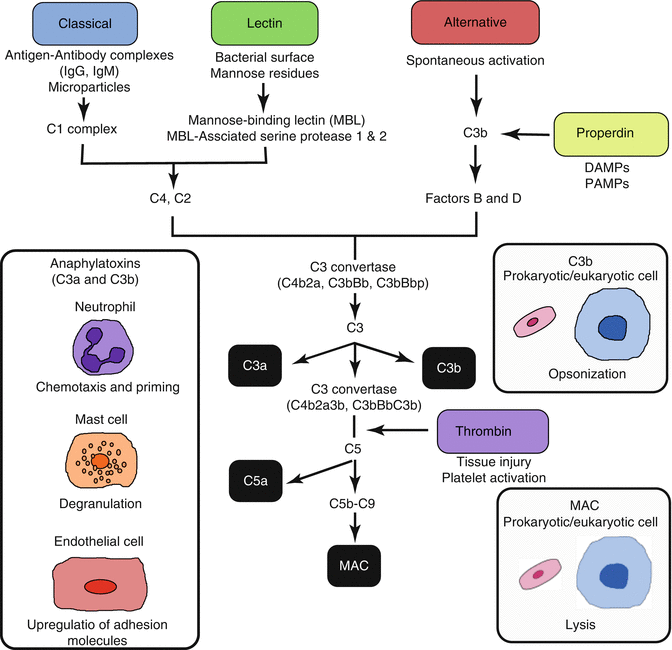
Fig. 12.2
Overview of complement activation pathways and biologic effects mediated by complement pathway products. MAC membrane attack complex
However, two additional complement activation pathways have been recently described—the properdin and thrombin pathways (Fig. 12.2). The properdin pathway is activated after recognition of DAMPs on foreign and apoptotic cells and allows for the assembly of C3 convertase on the cell surface [29]. DAMPs expressed following ischemia or tissue disruption implicate the properdin pathway as an important link between innate immunity and inflammation following shock. The fifth complement activation pathway is through the clotting factor thrombin. Thrombin may directly act as a C5 convertase linking both the complement and coagulation systems [30, 31].
Neutrophils are the predominant effector cells of the innate immune system, and the ultimate outcome of both the macrophage and complement response to shock is neutrophil recruitment to the ischemic/injured tissue. Moreover, recent data suggest that danger signals associated with ischemic or necrotic cells (nucleotide oligomerization domain receptors—NLRP3 inflammasome activation, intravascular gradient of chemokine C-X-C ligand 2, and signaling through N-formyl peptide receptor-1) guide neutrophils through healthy tissue to sites of sterile inflammation [32]. Neutrophils are highly regulated, effective phagocytes whose main function is to seek out and destroy pathogenic microorganisms. However, these same cellular and biochemical events in response to infection paradoxically contribute to severe inflammation and tissue injury in the setting of ischemia/reperfusion and tissue disruption.
Neutrophils contain four types of intracellular granules: azurophilic (primary), specific (secondary), gelatinase (tertiary) granules, and secretory vesicles that contribute to tissue injury [33]. Granule-associated proteins delivered to the neutrophil surface following activation facilitate cell adhesion and chemotaxis, and others aid in microbial killing or the pathologic response to shock. Neutrophil elastase is one such factor found in the azurophilic granules and is a serine protease which functions as part of the phagolysosome. However, once released extracellularly following pronounced inflammation, elastase stimulates the release of growth factors and proinflammatory cytokines, formation of reactive oxygen species, and cellular apoptosis [33–35]. Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases found in both specific and gelatinase granules and influence tissue remodeling, cell migration, and angiogenesis. The release of MMP-2 (gelatinase A) and MMP-9 (gelatinase B) from gelatinase granules contributes to tissue injury [36, 37]. However, MMP-8, which is released from the specific granules, may have anti-inflammatory properties through cleavage of neutrophil-derived cytokines and upregulation of anti-inflammatory cytokines [38]. α[alpha]-Defensins are released from the azurophilic granules and are cationic peptides that can associate with bacterial cell walls, form pores, and disrupt the cell wall integrity. α[alpha]-Defensins also trigger macrophage release of TNF-α[alpha] and interferon-γ[gamma] further amplifying the immune response [39]. Myeloperoxidase, a major constituent of primary granules, and NADPH oxidase are responsible for neutrophil generation and release of reactive oxygen species [40]. Reactive oxygen species disrupt intercellular tight junctions, increase endothelial permeability, and accelerate apoptosis. [41, 42]
There is ample evidence demonstrating the strong coupling of the effects of shock and inflammation. After an initial insult, neutrophils become activated and localize to the injured/ischemic tissue resulting in more localized inflammation. This response is often tolerated if the injury is limited. However, a second insult (i.e., infection, ischemia/reperfusion, or tissue injury) following trauma and shock may activate systemic primed neutrophils and incite an unbridled inflammatory response resulting in multiple organ failure and mortality [43]. Therefore, it is important to understand the pathophysiology of shock and the maladaptive immune response following severe injury.
Physiological Response to Shock
The pathophysiology of shock is complex, but the physiological response to shock is ultimately driven by tissue hypoperfusion. While this physiological response may differ depending on the etiology of shock, the initial priority is to preserve cerebral and coronary perfusion. The cardiovascular response to differing etiologies of shock is an illustration of this point, manifesting as variations in the systemic vascular resistance, central venous pressure, and cardiac index (Table 12.3).
Table 12.3
Hemodynamic responses to shock
Cardiac index | SVR | Venous capacitance | CVP/PCWP | SVO2 | |
|---|---|---|---|---|---|
Hemorrhagic | ↓ | ↑ | ↓ | ↓ | ↓ |
Traumatic | ↓ | ↑ | ↓ | ↓ | ↓ |
Cardiogenic | ↓ | ↑ | ↑ | ↑ | ↓ |
Neurogenic | ↓ | ↓ | Variable | Variable | ↑ |
Obstructive | ↓ | ↑ | ↑ | ↑ | ↓ |
Septic | ↑ | ↓ | Variable | Variable | ↑ |
The body has multiple, complex systems in place that regulate these responses and are sensitive to small decreases in blood pressure and pH and rapidly respond to correct these changes. Stretch receptors in the heart and baroreceptors located in the carotid sinuses and aortic arch sense decreases in blood pressure and respond by a reflexive activation of the sympathetic nervous system. The end result of sympathetic activation is an increase in peripheral vascular resistance and cardiac output through increased vasoconstriction, heart rate, and ventricular contractility [43]. Activation of the sympathetic nervous system further induces the release of catecholamines from the adrenals. The carotid and aortic bodies also detect changes in partial pressures of oxygen and carbon dioxide as well as arterial pH. Central chemoreceptors are also present in the medulla oblongata, which primarily detect changes in pH within the cerebrospinal fluid but have also been shown to respond to hypercapnia and hypoxia. Activation of these receptors results in an increase in respiratory rate as well as peripheral vascular resistance with a corresponding decrease in heart rate, which is known as the Cushing reflex [44]. The renin-angiotensin-aldosterone system also responds to decreases in blood pressure. The kidney’s macula densa senses a decrease in blood pressure and signals the juxtaglomerular cells to secrete renin. Plasma renin is then responsible for the conversion of angiotensinogen to angiotensin I in the liver. Subsequently, angiotensin I is primarily converted to angiotensin II in the lungs. Angiotensin II is a potent vasoactive peptide, which increases blood pressure through vasoconstriction and also stimulates the adrenal cortex to secrete aldosterone. The role of aldosterone is to promote sodium reabsorption in the distal tubules and collecting ducts of the nephron, thus expanding intravascular volume and subsequently increasing blood pressure [44].
Other hormones besides aldosterone are also involved in physiological responses to shock. Hypoperfusion activates the hypothalamic-pituitary-adrenal axis stimulating the hypothalamus to release corticotropin-releasing hormone and, subsequently, release of adrenocorticotropic hormone (ACTH) from the anterior pituitary. ACTH stimulates the adrenal cortex to release cortisol, which promotes a catabolic state as well as kidney reabsorption of sodium. The posterior pituitary releases antidiuretic hormone (ADH) in response to both hypovolemia and increased plasma osmolality, which increases water permeability in the distal tubule and collecting duct of the kidney, resulting in an increase in intravascular volume. ADH, which was first described as arginine vasopressin, acts a peripheral vasoconstrictor, shunting blood from splanchnic organs to the cerebral and coronary circulation [44].
Categories of Shock
Classically, four categories of shock were described by Blalock based on hemodynamic profiles, which are still useful today: hypovolemic, cardiogenic, distributive, and neurogenic shock [45]. However, clinicians who manage trauma patients prefer to add two additional categories due to their unique pathophysiology: traumatic and obstructive shock (Table 12.1). Treatment of all forms of shock is empiric until the etiology can be determined. Initially, a secure airway, to ensure adequate oxygenation (pO2) and ventilation (pCO2), and adequate intravenous access must be established, with the initiation of volume restoration. Subsequently, a systematic physical exam and appropriate diagnostic studies are performed to identify the cause(s) of shock.
Hemorrhagic Shock
In trauma patients, loss of circulating volume from hemorrhage is the most common etiology of shock. Therefore, hypotension in the trauma patient should be presumed to be from hemorrhage until proven otherwise. The physiological response and clinical presentation of hemorrhagic shock have been characterized according to the volume of blood loss (Table 12.4). Blood loss of less than 15 % of the total circulating blood volume results in very few clinical symptoms and may be overlooked. Mild symptoms of tachycardia and anxiety are usually evident in patients with up to 30 % blood loss. However, trauma patients usually do not become hypotensive, confused, or develop significant tachycardia until a blood loss of greater than 30 %. If trauma patients lose greater than 40 % of their circulating blood volume, they are usually obtunded on presentation with severe hypotension and tachycardia and are at significant risk for death. Young patients, with greater physiological reserve and stronger compensatory mechanisms, may tolerate larger volumes of blood loss while exhibiting fewer clinical signs. On the other hand, elderly patients, due to their preexisting diseases or medications to manage comorbidities, may be at a greater risk for bleeding (warfarin, platelet inhibitors, and direct thrombin inhibitors) and inability to compensate for hypovolemia (antihypertensive agents and β[beta]-blockers).
Table 12.4
Classification of hemorrhagic shock
Class I | Class II | Class III | Class IV | |
|---|---|---|---|---|
Blood loss (mL) | Up to 750 | 750–1,500 | 1,500–2,000 | >2,000 |
Blood loss (% BV) | Up to 15 % | 15–30 % | 30–40 % | >40 % |
Heart rate (beats/min) | <100 | >100 | >120 | >140 |
Blood pressure | Normal | Orthostatic | Decreased | Severely decreased |
Pulse pressure | Normal | Narrowed | Narrowed | Very narrow or unobtainable |
Hourly urine output | >0.5 mL/kg | >0.5 mL/kg | <0.5 mL/kg | Minimal |
Mental status | Normal | Anxious | Confused | Obtunded |
It is important to identify the source of bleeding in patients with hemorrhagic shock following trauma, since controlling the hemorrhage is imperative to resuscitate the patient. The potential sites capable of large volume blood loss include external, intrathoracic, intra-abdominal, retroperitoneal spaces, as well as long bone and pelvic fractures. These can often be rapidly diagnosed based on a focused physical exam besides ultrasound (FAST exam) or plain radiographs. Treatment consists of controlling the blood loss and commencement of intravenous volume resuscitation.
Resuscitation strategies may vary by institution and region but continue to evolve as the pathophysiology of hemorrhagic shock, and how it relates to inflammation and coagulation, is better understood. There remains much controversy behind resuscitation strategies, and two of these proposed strategies include hypotensive resuscitation and the minimization of dilutional coagulopathy. However, a balance must exist to ensure adequate perfusion in order to minimize the post-resuscitation inflammatory response and further coagulopathy. A recent management strategy in the severely injured trauma patient is a combination of these strategies known as damage control resuscitation [46]. The principal components are (1) to employ “hypotensive resuscitation” to a systolic blood pressure of 80–90 mmHg in order to minimize ongoing hemorrhage and clot disruption, (2) to minimize dilutional coagulopathy by limiting crystalloid transfusion, and (3) to administer preemptive blood components to replace whole-blood loss. While this strategy has been reported to improve survival, the benefits of the individual components remain to be established [47]. However, hypotensive resuscitation should not be employed in traumatic brain injury patients, in which systolic blood pressures should be maintained greater than 110 mmHg or greater than 120 in settings without invasive intracranial monitoring capabilities, in order to sustain cerebral perfusion pressures and minimize secondary insults [48]. Closed head injuries are at increased risk for disturbances in autoregulation and elevated intracranial pressure, thus lowering cerebral perfusion. If the cerebral perfusion pressure falls below 50 mmHg, patients are at high risk for ischemic brain injuries, and therefore cerebral perfusion pressure should be maintained between 50 and 70 mmHg [49]. Attempts to maintain cerebral perfusion pressure greater than 70 mmHg have been associated with an increased risk of ARDS without any improvement in neurological outcomes [50].
The optimal initial resuscitation fluid given to patients in hemorrhagic shock continues to be debated. Crystalloids are the standard of care in the United States. Colloid fluids, such as albumin, dextran, gelatin, or hydroxyethyl starch, are hypothesized to be optimal fluids since they are retained in the intravascular plasma volume. However, to date, no randomized controlled trials have shown a survival benefit of colloid solutions compared to crystalloid solutions. The administration of human albumin has been extensively studied for over two decades without any improved mortality and is significantly more expensive than crystalloids [51, 52]. The starch-containing colloids are currently used in Europe but have been associated with nephrotoxicity and increased bleeding, and no randomized trials have demonstrated convincing improved outcomes [53, 54]. The same holds true for hypertonic saline. Although hypertonic saline (HTS) is a plasma volume expander and has immunomodulatory effects, a recent randomized trial failed to show improved survival. It is postulated that HTS may have delayed blood product transfusion or increased coagulopathy, leading to increased mortality [55]. Ultimately, intravascular volume should be restored with blood products as early as possible in patients with persistent hemorrhagic shock [56].
The endpoints of resuscitation remain unclear and extend beyond the normalization of blood pressure, heart rate, and urine output. Even when these parameters normalize, up to 85 % of severely injured patients still have evidence of tissue hypoperfusion reflecting compensated shock [57, 58]. Therefore, other parameters have been proposed including supranormal hemodynamic parameters (cardiac index, O2 delivery, and O2 consumption), mixed venous oxygen saturation (SvO2), arterial base deficit and lactate, gastric tonometry, and near-infrared spectroscopy (NIRS). Although achieving supranormal hemodynamic parameters (cardiac index >4.5 L/min/m, O2 delivery >600 mL/min/m2, and O2 consumption >170 mL/min/m2) were proposed to improve survival and decrease of MOF, no adequately powered randomized prospective trials demonstrated improved outcomes [59–62]. Many studies showed no difference in outcomes and suggested that the ability of patients to achieve these parameters was predictive of survival, rather than the supranormal endpoints themselves [63–65]. In fact, recent data indicate supranormal resuscitation results in excessive crystalloid infusion, abdominal compartment syndrome, MOF, and mortality [66, 67]. Although SvO2 should reflect the adequacy of O2 delivery to tissues, resuscitating critically ill patients to normal SvO2 (>70 %) did not improve survival or MOF [68]. Both arterial base deficit and lactate levels reflect the degree of tissue ischemia and are proportional to the depth and severity of shock. Although several studies have shown that initial values and time to normalization are predictive of transfusions, MOF, and mortality, it has not been proven that normalizing base deficit or lactate levels as an endpoint for resuscitation improves survival [60, 69–72]. Gastric tonometry allows for the detection of subclinical ischemia through the measurement of gastric pCO2 and calculated intramucosal pH (pHi). The difference between intragastric pCO2/pHi and arterial pCO2 correlates with the degree of gastric ischemia and is predictive of MOF and mortality in trauma patients [73–75]. However, measurement requires withholding gastric feeding and suppressing gastric acid secretion, and normalization of pHi or pCO2 gap as endpoints for resuscitation has not been shown to improve outcomes. NIRS can simultaneously measure tissue pO2, pCO2, and pH, and preclinical studies have suggested that measurement of tissue pO2, pCO2, and pH of solid organs and skeletal muscle may be a better predictor of outcomes and provide better endpoints for resuscitation [76–78]. NIRS-derived tissue oxygenation saturation has also been found to predict the need for blood transfusion in trauma patients who initially appeared hemodynamically stable [79]. Although preliminary, and no studies showing improved outcomes with NIRS resuscitation endpoints have been performed, this technology provides clinicians a tool to quickly assess tissue oxygenation in a noninvasive manner and shows promise to guide resuscitation as well as triage patients in the field.
Traumatic Shock
Traumatic shock is a variant of hemorrhagic shock, combining the effects of tissue injury and long bone fractures with substantial blood loss. Traumatic shock initiates a massive proinflammatory response that increases the systemic inflammatory response syndrome (SIRS) and risk for acute respiratory distress syndrome (ARDS) and multiple organ failure (MOF), which rarely occur following simple hemorrhagic shock [80]. In severely injured trauma patients, the inflammatory response occurs within 30 min following injury, and this early increase in cytokines is associated with worse outcomes [81, 82]. This magnified inflammatory response may be due to the combination of both ischemia and direct cellular injury leading to a greater release of endogenous molecules, called DAMPs or alarmins, which interact with cells of the innate immune system and initiate an inflammatory response (Fig. 12.3) [16, 83, 84]. Mechanical cell rupture from trauma, or ischemia-induced apoptosis, leads to the passive release of intracellular contents into the extracellular environment, which contain DAMPs [85]. High-mobility group box (HMGB) 1, heat shock proteins (HSPs), and mitochondrial DNA have been reported to be elevated following trauma, and plasma HMGB1 levels correlate well with injury severity score and base deficit [86–88]. S100 proteins are calcium-binding proteins which are found primarily in oligodendrocytes, astrocytes, and Schwann cells and act as ligands for the transmembrane receptor for advanced glycation endproducts (RAGE), which have direct effects on the innate immune system and inflammation [89]. Elevated plasma levels of S100B have been identified in traumatic brain injury patients and correlated well with poor outcomes [90, 91]. Secretory RAGE (sRAGE) is cleaved from RAGE and is elevated in the plasma within 30 min following injury. High levels have been associated with coagulopathy and complement activation in trauma patients [92, 93]. Furthermore, cell surface saccharides, heparan sulfate and hyaluronic acid, act as ligands for TLR4 and have been linked to SIRS [94]. The net result of these events is a “genomic storm,” resulting in a dramatic increase in both proinflammatory and anti-inflammatory signaling [95]. Treatment of traumatic shock focuses on minimizing the immune dysregulation via prompt control of hemorrhage, adequate volume resuscitation, restoring coagulation capacity, debridement of nonviable tissue, and stabilization of fractures.
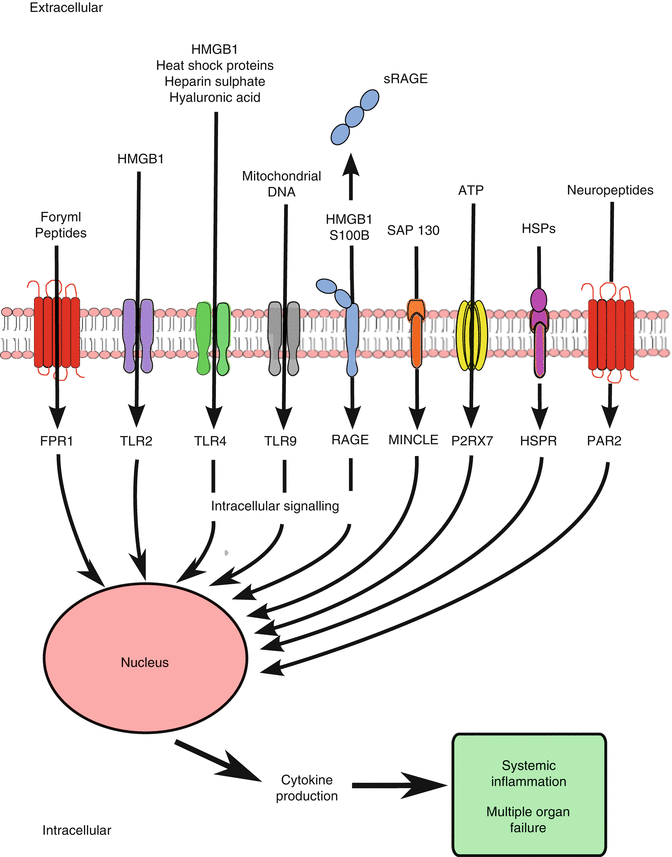
Fig. 12.3
DAMPs or alarmins resulting from trauma and shock and their receptors for proinflammatory immune activation. HMGB high-mobility group box protein, (s)RAGE (secretory) receptor for advanced glycation endproducts, ATP adenosine 5′-triphosphate, HSP(R) heat shock protein (receptor), FPR formyl peptide receptor, TLR Toll-like receptor, Mincle macrophage-inducible C-type lectin, P2RX7 purinergic receptor, PAR proteinase-activated receptor, NF nuclear factor
Distributive Shock
Distributive shock is characterized by an overall decrease in systemic vascular resistance from the failure of vascular smooth muscle contraction, which results in hypotension and poor tissue perfusion. There are multiple etiologies for distributive shock (Table 12.5), but all result in endothelial and vascular dysfunction from exogenous and endogenous inflammatory mediators or as a response from prolonged and severe hypoperfusion [96]. Ultimately, all forms of untreated and prolonged shock result in distributive shock.
Table 12.5
Causes of distributive shock
Sepsis |
Noninfectious |
Pancreatitis |
Burns |
Adrenal insufficiency |
Anaphylaxis |
Prolonged and severe hypotension |
Hypovolemic shock |
Cardiogenic shock |
Obstructive shock |
Metabolic |
Lactic acidosis |
Carbon monoxide poisoning |
The most common etiology for distributive shock is severe sepsis, which is increasing in incidence and kills approximately a third of those diagnosed in the United States [97, 98]. However, distributive shock can occur with advanced traumatic shock [96]. Most etiologies have a common pathway involving the production of nitric oxide (NO). NO is synthesized by nitric oxide synthase, which converts l-arginine to NO and l-citrulline. NO may then act locally or diffuse into nearby cells where it activates cytosolic guanylate cyclase forming cyclic GMP (cGMP). In the vascular smooth muscle, increases in cGMP result in smooth muscle relaxation and vasodilation. There are currently three recognized isoforms of NOS in eukaryotes: endothelial NOS (eNOS) and neuronal NOS (nNOS), which are calcium dependent and constitutively expressed, and the inducible isoform (iNOS), which is calcium independent, and its expression is induced by cytokines and inflammatory mediators [99]. Normally, NO production through eNOS regulates microvascular homeostasis and regulation of blood flow. However, in sepsis and inflammation induced by protracted shock, inflammatory stimuli (LPS, IL-1β[beta], macrophage migration inhibitory factor [MIF], and IL-6) induce iNOS expression and the creation of large amounts of NO (Fig. 12.4) [99]. This results in systemic vasodilation, hypotension, early neutrophil activation, decreased immune cell function (monocyte/macrophages, dendritic cells, T-lymphocytes, and late neutrophil function), and ultimately organ dysfunction.
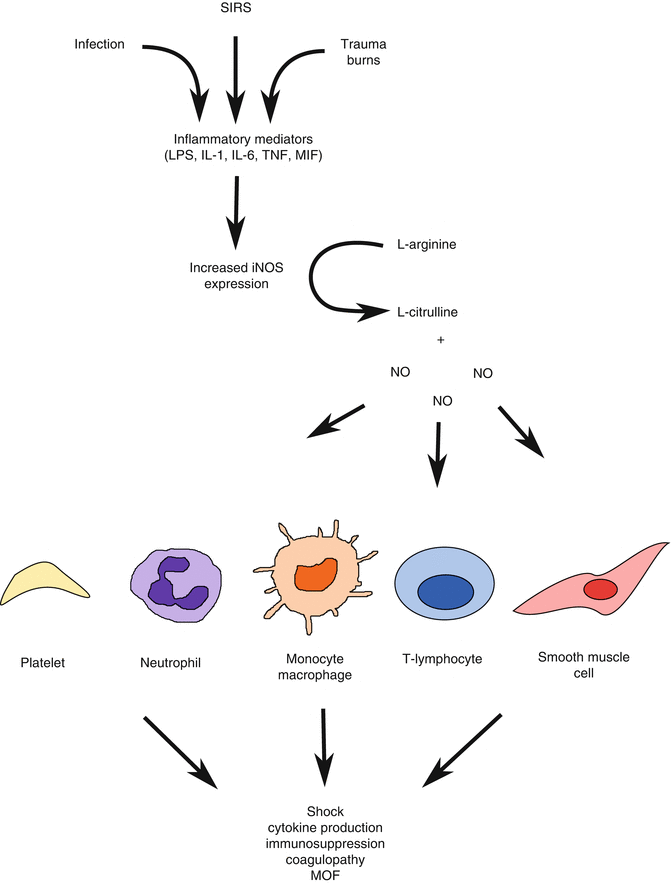
Fig. 12.4
Role of NO in the pathophysiology of shock following trauma, systemic inflammatory response syndrome (SIRS), and infection
Early goal-directed resuscitation for patients in septic shock has been shown to improve survival [100]. Within the first 6 h after the diagnosis of septic shock, the goal of resuscitation is to achieve a central venous pressure of 8–12 mmHg, mean arterial pressure greater than or equal to 65 mmHg, urine output greater than or equal to 0.5 ml/kg/h, and a central venous or mixed venous oxygen saturation greater than or equal to 70 % or 65 %, respectively [101]. Vasopressors may be needed to maintain the mean arterial pressure greater than or equal to 65 mmHg, and norepinephrine is the initial vasopressor of choice [101]. Vasopressin may be subsequently added to norepinephrine, and dobutamine may be used in patients with myocardial dysfunction for inotropic support. Empiric antibiotics must be started as soon as the diagnosis is suspected and chosen based on the most likely pathogens. The use of steroids remains controversial but should be considered in patients when hypotension responds poorly to adequate fluid resuscitation and vasopressors [101].
Neurogenic Shock
Acute spinal cord injury (SCI) above the level of T6 has been associated with a higher incidence of neurogenic shock [102, 103]. In developed countries, SCI has an incidence of about 50 cases/million population per year, but less than 20 % results in neurogenic shock [104, 105]. Neurogenic shock occurs secondary to spinal cord injuries usually from vertebral body fractures of the cervical or high thoracic spine, which disrupt the sympathetic regulation of the cardiovascular system. It may also result from compression of the spinal cord from an epidural hematoma. Although, often considered a form of distributive shock, the pathogenesis of neurogenic shock is unique in that shock results from the inhibition of multiple systems to regulate blood pressure. This occurs from disruption of the descending medullary basal sympathetic tone to the sympathetic preganglionic vasomotor neurons in the thoracolumbar cord, which affects four organs: (1) the heart, resulting in bradycardia and decreased inotropic function; (2) the peripheral vascular system, increasing systemic vasodilation; (3) the adrenal medulla, decreasing circulating epinephrine and norepinephrine; and (4) the kidney, decreasing stimulation of the juxtaglomerular cells which activate the renin-angiotensin-aldosterone axis [106]. It is important to note that neurogenic shock is a different entity than spinal shock. Although spinal shock often accompanies neurogenic shock, spinal shock does not refer to circulatory collapse and is characterized by a marked reduction or abolition of somatic and/or reflex functions of the spinal cord caudal to the injury site (loss of sensation accompanied by motor paralysis and areflexia/hyporeflexia). On the other hand, neurogenic shock is characterized by hypotension following injury to the central nervous system [102].
Diagnosing neurogenic shock can be difficult in patients with multisystem trauma, and hemorrhagic shock must first be ruled out. Neurogenic shock should be suspected in patients with hypotension (SBP <90 mmHg), relative bradycardia, warm extremities, motor and/or sensory deficits suggestive of a spinal cord injury, and radiographic evidence of vertebral column fractures. Spinal cord injuries can later be defined by an MRI once the patient is hemodynamically stable. Initial treatment consists of securing an airway. If the patient requires endotracheal intubation, it should be performed by experienced personnel, with standard precautions, including in-line immobilization. Subsequent interventions should be focused on expanding intravascular volume to maintain a systolic blood pressure greater than 90 mmHg or a cerebral perfusion pressure greater than 50 [107–109]. It is imperative to correct the hypotension rapidly since decreased spinal cord perfusion is detrimental to the initial SCI and perpetuates a secondary injury [110]. If hypotension persists after adequate volume has been given, vasopressors and inotropic agents should be used. In the case of decreased peripheral vascular resistance and adequate cardiac output and heart rate, norepinephrine is preferred. In the unusual situation where cardiac output and heart rate are the dominant factors, dopamine may be optimal [110].
Cardiogenic Shock
Clinically, cardiogenic shock is defined by tissue hypoxia secondary to myocardial dysfunction in the presence of adequate intravascular volume. It is characterized by a systolic blood pressure of less than 90 mmHg for at least 1 h that is (1) not responsive to fluid administration alone and (2) associated with a cardiac index of less than 2.2 L/min/m2 in the setting of a pulmonary capillary wedge pressure (PCWP) greater than 18 mmHg [111]. The leading cause of cardiogenic shock is left ventricular failure secondary to acute myocardial infarction [112]. However, there are multiple potential etiologies of cardiogenic shock following trauma (acute myocardial infarction, cardiomyopathy, valvular heart disease, arrhythmias, myocardial injury), but all ultimately result in cardiac ischemia, leading to cardiac dysfunction and, thereby, promoting further ischemia resulting in a vicious cycle. Consequently, mortality from cardiogenic shock can be as high as 80 % [113]. Once a critical mass of ischemic/necrotic left ventricular myocardium reaches approximately 40 %, there is a life-threatening decrease in pumping capability, stroke volume, and cardiac output [111].
In the trauma patient, cardiogenic shock needs to be considered even in the setting of hemorrhagic shock, and a rapid physical assessment should be performed in the emergency department. Patients in cardiogenic shock will usually present with hypotension and jugular venous distension. The presence of a cardiac murmur, ascites, or peripheral edema suggests a preexisting cardiac problem. The diagnosis can be further pursued with an ECG, laboratory values (elevated troponin and BNP), chest radiograph, and echocardiography. The initial step in management should be to ensure an adequate airway. If the likely etiology is an acute myocardial infarction, prompt revascularization is the only intervention that has consistently been shown to reduce mortality in patients with cardiogenic shock [112]. Fibrinolytic therapy should be avoided in the trauma patient and is not as effective once cardiogenic shock has developed [114]. If hypotension is unresponsive to fluid challenges, vasopressors should be used. Norepinephrine and dopamine are considered first-line drugs of choice in this situation. If patients continue to be nonresponsive to pharmacologic therapy, devices, such as intra-aortic balloon pumps or left ventricular assist devices, may be required to improve cardiac perfusion and allow time for the myocardium to recover.
Extracardiac Obstructive Shock
Extracardiac obstructive shock results from an acute obstruction to circulatory flow by either impaired diastolic filling of the right ventricle (tension pneumothorax or cardiac tamponade) or obstruction of right ventricular output (massive bronchovenous air embolism/pulmonary embolism or pulmonary hypertension). There are several etiologies of obstructive shock, which involve IVC obstruction/compression or increases in intrathoracic/intracardiac pressures, but tension pneumothorax and cardiac tamponade are the most common following trauma.
The rapid diagnosis of a tension pneumothorax is imperative and should be made on clinical examination. Clinical findings include hypotension, respiratory distress, diminished breath sounds over one hemithorax, jugular venous distention, and tracheal deviation. A pneumothorax develops secondary to a breach in the visceral, parietal, or mediastinal pleura, and in a tension pneumothorax, the pleural defect functions as a one-way valve with air entering the pleural cavity on inspiration but is unable to exit on expiration [115]. This leads to an increase in intrathoracic pressure and a decrease in venous return to the heart, effectively reducing cardiac output and resulting in shock. Empiric treatment is pleural decompression with a tube thoracostomy; however, if a chest tube is not readily accessible, as in the prehospital setting, decompression with a large-caliber needle may be performed as a temporizing measure. Following pleural decompression, there should be a rapid resolution of the hypotension and improvement in respiratory parameters.
Cardiac tamponade results from the accumulation of fluid within the pericardial sac. In trauma, cardiac tamponade is seen in both penetrating and blunt trauma scenarios in which the myocardium or vessels are injured and blood acutely fills the pericardial space, increasing the intracardiac pressures. This decreases right atrial filling and, ultimately, compresses the right ventricle, resulting in a significant reduction in cardiac output. Clinical findings may include those of Beck’s triad (hypotension, muffled heart tones, and jugular venous distention), but this may be absent in acute tamponade. The diagnosis can be confirmed by a rapid bedside ultrasound, demonstrating pericardial fluid, right atrial collapse, and poor distensibility of the right ventricle. Treatment consists of an emergent pericardiocentesis with placement of a catheter to confirm the diagnosis and to relieve the tamponade. If the patient is in circulatory arrest, an emergent left thoracotomy for pericardial decompression should be performed. Patients may benefit from a fluid bolus if hypovolemic, but in cases of normovolemia or hypervolemia, fluid administration may be deleterious resulting in pulmonary eduma [116]. Isoproterenol may also be beneficial until pericardial decompression can be performed. Isoproterenol increases heart rate and cardiac output and decreases right atrial pressure and systemic vascular resistance [117].
Bronchovenous air embolism is another cause of right ventricular outflow obstruction and can occur following blunt or penetrating thoracic trauma with damage to the great veins or air entry through disrupted smaller venous vessels. The incidence following thoracic trauma has been estimated at 4–14 %, with mortality as high as 80 % in blunt trauma patients [118, 119]. Bronchovenous air embolism may be diagnosed by CT or echocardiography. The initial management of venous air embolism is to immediately give the patient 100 % supplemental oxygen and to place the patient in Trendelenburg with left lateral decubitus positioning. This may prevent air from traveling through the right side of the heart into the pulmonary arteries. A central venous catheter may then be placed into the right atrium and aspiration performed to remove the air from the venous circulation. Hyperbaric oxygen therapy may also be considered as a possible therapy in symptomatic patients.
Coagulopathy
Trauma continues to be the leading cause of morbidity and mortality worldwide with road traffic injuries, self-inflicted injuries, and interpersonal violence accounting for the three leading causes of death in persons aged 15 through 44 years [120]. Uncontrolled hemorrhage is the second leading cause of death in trauma patients and accounts for nearly 40 % of early mortality, which is preceded only by central nervous system injury [121]. Moreover, approximately one-third of trauma patients have a coagulopathy on arrival to the emergency department, further contributing to hemorrhage, and these patients are four times more likely to die than those without a coagulopathy [122–124].
Coagulopathy, or post-injury hemorrhage that persists despite control of surgical bleeding, was originally described over 60 years ago and has been referred to by many names: medical bleeding, diffuse bleeding diathesis, posttransfusion bleeding disorder, medical oozing, and disseminated intravascular coagulation [125]. Clinically, the coagulopathy manifests as nonsurgical bleeding from mucosal lesions, serosal surfaces, wounds, and vascular access sites, which continues even after control of identifiable vascular bleeding. Although post-injury coagulopathy has long been recognized, the pathophysiology remains poorly understood, and both clinical and laboratory parameters remain feeble predictors of coagulopathy. In 1982, our group described the “bloody vicious cycle,” also referred to as “the lethal triad,” in which the synergistic effects of acidosis, hypothermia, and coagulopathy combined create an irreversible clinical deterioration in patients receiving massive transfusions resulting in death by exsanguination despite surgical control of bleeding (Fig. 12.5) [126]. This was later correlated with decreased clotting factor concentrations and a corresponding prolongation of traditional measures of coagulopathy, such as prothrombin time (PT) and activated partial thromboplastin time (aPTT) suggesting either a consumption or dilution of clotting factors [127]. In spite of this, development of coagulopathy following massive transfusion still could not be adequately predicted with clinical and laboratory parameters [128]. However, recent evidence suggests an endogenous coagulopathy associated with severe trauma, termed the acute coagulopathy of trauma (ACOT), which occurs early and is independent of the secondary effects of body temperature, acidosis, and clotting factor levels [129].
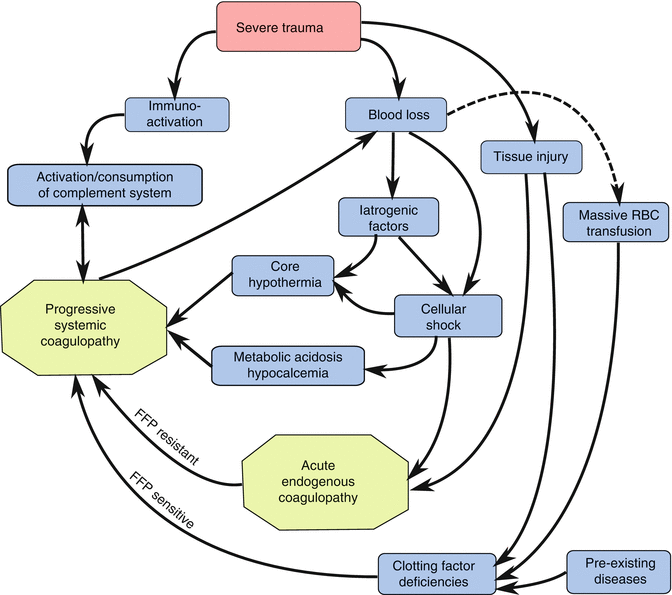
Fig. 12.5
The “bloody vicious cycle” of the acute coagulopathy of trauma. This updated cycle incorporates both the early acute endogenous coagulopathy of trauma, which is resistant to clotting factor replacement with fresh frozen plasma (FFP resistant), and a subsequent secondary coagulopathy that may be due to hypothermia, acidosis, clotting factor deficiency (FFP sensitive), or any combination of factors
Cell-Based Model of Coagulation
Effective management of post-injury coagulopathy requires a fundamental understanding of the coagulation process, which depends on an intricate balance between the anticoagulant, procoagulant, and fibrinolytic systems. Major proteins involved in these systems are listed in Table 12.6. Traditionally, coagulation was described as a cascade model composed of a sequential series of steps in which activation of one clotting factor led to the activation of another, resulting in thrombin generation. This model reflects two distinct pathways, the intrinsic and extrinsic pathways, which converge on a common pathway with the conversion of factor X to factor Xa. Although this model improved our understanding of key physiological events in vivo, it was clear from early studies that cells were important participants in coagulation. Hemostasis is not possible without platelets, and tissue factor (TF) is an integral membrane protein. However, cells were viewed only as donors of an anionic phospholipid surface for procoagulant complex assembly, and this model was supported by traditional plasma-based laboratory tests of isolated coagulation in a test tube, which do not correlate with clinical hemostasis.
Table 12.6
Proteins involved in coagulation, anticoagulation, and fibrinolysis
Protein | Source | Activated by | Function |
|---|---|---|---|
Procoagulant | |||
Tissue factor | Subendothelium, monocytes in response to IL-6 | Exposure to circulating platelets | Complex with VII to initiate clot formation |
Fibrinogen (factor I) | Liver, activated platelets | Thrombin | Clot formation |
Factor V | Liver, activated platelets | Thrombin | Cofactor that accelerates conversion of prothrombin to thrombin; Leiden mutation renders it resistant to inactivation by APC |
Factor VII | Liver | Thrombin, Xa, XIa, XIIa | Complexes with TF to convert X to Xa |
rFVIIa | N/A | N/A | Complexes with TF → activate X |
Binds to activated plts → activate X (bypassing VIII and IX) | |||
Activate TAFI | |||
Factor VIII | Endothelial cell | Thrombin | Cofactor, activates factor X |
Liver sinusoidal cells | |||
Factor IX | Liver | Factor XI | Cofactor, activates factor X |
Factor X | Liver | Factor VIII, factor IX | Converts prothrombin to thrombin |
Factor XI | Liver | Thrombin | Activates IX |
Factor XIII | Liver | Thrombin | Cross-links fibrin |
Anticoagulants | |||
Heparin sulfates | Endothelial cells | Ischemia, hypoxia | Activation of ATIII |
Antithrombin III | Liver | Thrombin | Inhibition of thrombin, Xa, Xia, XIIa |
Protein C | Liver | Thrombin-TM-EPCR complex | Irreversibly inactivates Va and VIIIa |
Protein S | Liver | Cofactor for protein C | |
Thrombomodulin (TM) | Endothelial cell
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|




