Fig. 16.1
Interaction between sarcopenia and diabetes. Insulin resistance in skeletal muscle is the most important link between sarcopenia and diabetes. Sarcopenia and obesity appear to have additive effects on insulin resistance and age-related changing in body composition. Diabetes, sarcopenia, and obesity may provide a vicious cycle: loss of skeletal muscle mass affects glucose disposal and energy homeostasis impairment affects muscle protein content. Coexistence of sarcopenia with diabetes or/and obesity increases the risk for fall or disability and cardio- and cerebrovascular diseases
16.3 Evidences in Basic Studies for Sarcopenia and Diabetes
16.3.1 Muscle Fiber-Type Switching
Evaluation of changes in skeletal muscle fiber composition during the early stages of the metabolic syndrome and diabetes is required to provide essential insights as to fiber-type distribution. Age-related sarcopenia is associated with muscle constitution in differing amounts, in individual skeletal muscle [24], and a decrease of size and number of fast type II muscle fiber has been observed [25, 26]. The EDL, or the extensor digitorum longus glycolytic muscle, is predominantly composed of type IIb fibers which has mainly reduced by 25–30 % in the cross-sectional area of the skeletal muscle of humans aged 70 [27]. In addition, insulin resistance is associated with higher proportions of glycolytic fast-twitch type IIb fibers [28]. The muscle transformation studies of the extended periods of bed rest have observed the slow-to-fast muscle transitions (myosin heavy chain, MyHCI to MyHCII) [29]. This shift in fiber-type composition may lead to the increase of the total EDL content in the aging process of muscle atrophy; however, this increase failed to prevent natural muscle aging and resulted in total skeletal muscle mass reduction in sarcopenia. The decrease in total type II fiber may have negative effects on the production of essentially muscular power in daily life, which is a pathognomonic change in sarcopenia or sarcopenia in diabetes.
16.3.2 Inflammation in Skeletal Muscle
Inflammation, an important mediator in the pathogenesis of insulin resistance, has been observed in both diabetes mellitus and sarcopenia. Since the muscle is the primary tissue which produces and responds to a variety of hormones and cytokines, it has been involved in modulating muscle protein degradation and myogenesis through the prevention of inflammation; further, pro-inflammation is counteracted by insulin resistance [30]. TNF-α is highly expressed in adipose tissues from obese subjects, which develops insulin resistance, induces IL-6 [31], and blocks muscle tissues differentiation leading to sarcopenia [32]. In addition, IL-6 and TNF-α activate TNF-α-related apoptosis-inducing ligand receptors (TNFR1) which stimulate inhibitor of κB kinase [33, 34], causing NF-κB activation resulting in processing protein degradation in the muscle. The effects of insulin antagonizing mediated by IL-6 on skeletal muscle have been described and chronic exposure to IL-6 caused inflammation which impaired insulin-stimulated GLUT4 translocation in skeletal muscle [35]. STAT3, signal transducer and activator of transcription 3, has been characterized in myokines signaling [36] which can link the activation of signal transducers of the Janus kinase (JAK) protein [37] and increased activity of the transcription factor C-EBPß and C-EBPδ involved in muscle wasting [38] through ActII receptor and myostatin [39] (Fig. 16.2).
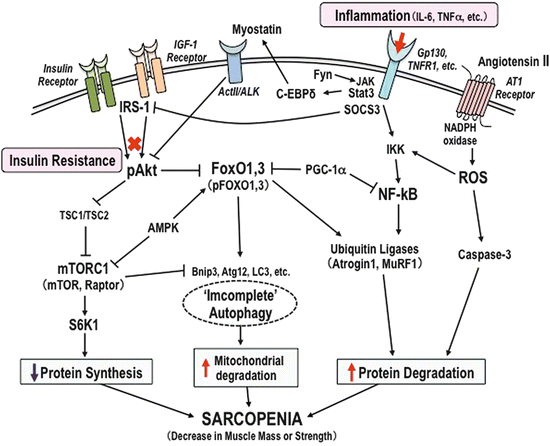
Fig. 16.2
Molecular mechanism of muscle wasting in diabetes. Decline of skeletal muscle mass or strength occurs under a variety of conditions and involves the regulation of muscle protein catabolism and mitochondrial dysfunction: FoxO1 and FoxO3 induce a decrease in muscle mass associated with an upregulation of ubiquitin E3 ligases MAFbx/atrogin-1 and MuRF1 expression. In cancer cachexia and sepsis, FoxOs inhibit the MAFbx/atrogin-1, MuRF1, and Bnip3 mRNA expression, which has been associated with inhibition of muscle fiber atrophy. NF-kB induces muscle breakdown by promoting protein degradations in skeletal muscle by regulation of ubiquitin-proteasome pathway (UPS). AGEs and angiotensin II via the angiotensin type 1 (AT1) receptor activate NADPH oxidase and lead to ROS production, facilitating activation of caspases, which contributes to muscle mass loss. IL-6 and TNF-α activate TNF-α-related apoptosis-inducing ligand receptors (TNFR1) which stimulate inhibitor of κB kinase (IKK) and cause NF-κB activation, thus processing the protein degradations in muscle. Signal transducer and activator of transcription 3 (STAT3) links the activation of signal transducers of the Janus kinase (JAK) protein and increases activity of the transcription factor C-EBPß and C-EBPδ involved in muscle wasting through ActII receptor and myostatin. Suppressor of cytokine signaling (SOCS3) targets IRS-1 in inflammation-induced insulin resistance. IRS-1 is rapidly degraded after IGF-1 stimulation and blocks FoxO1, leading to inhibition of atrophy. IRS-1 phosphorylation links to dephosphorylation of Akt, which mediates insulin resistance. Inactivation of tuberous sclerosis complex 1/2 (TSC1/TSC2) stimulates Akt–mTOR signaling and leads to “incomplete” autophagy in muscle wasting. AMPK inactivates mTOR and decreases the activation of ribosomal protein S6 kinase 1 (S6K1), an activator of protein synthesis, thereby increasing the rates of muscle mass loss. The autophagic regulator proteins, Atg12, LC3-II, and apoptotic genes Bnip3, promote mitochondrial disruption by activation of FoxOs during muscle atrophy
On the other hand, IL-6 or IL-15 [40, 41], which is secreted from skeletal muscle, suppresses TNF-α effects on the exercise conditions [42]. These cytokines produced from skeletal muscle are called “myokines” and exert autocrine, paracrine, or endocrine effects. The interaction between myokines production and diabetes is still unclear, but myokines can be the candidate biomarkers for metabolic disorders, including diabetes.
16.3.3 Molecule-Related Signaling Pathways in Muscle Wasting
Many reports have shown the molecular mechanisms for muscle wasting using animal models which fed on high-fat or sucrose diet and of diabetes. The molecules signaling pathways related to muscle wasting has been shown in this section.
16.3.3.1 Akt/PKB Signaling
In skeletal muscle, Akt/PKB plays a key role in insulin and PI3K/Akt/mTOR signaling pathway, which is regulating energy metabolism and protein synthesis. Previous studies have shown that SOCS3, suppressor of cytokine signaling, targeted to IRS-1 in inflammation-induced insulin resistance [43]. IRS-1 is rapidly degraded after IGF-1 stimulation and blocking of FoxO1 leads to inhibition of atrophy. The activation of Akt/PKB results in the glucose uptake through the stimulation and translocation of GLUT4, leading to the increased uptake of glucose, thus decreasing the amount of circulating glucose and regulating glucose metabolism upon insulin exposure [44, 45]. However, IRS-1 phosphorylation is linked to dephosphorylation of Akt/PKB [46], which mediates insulin resistance and further inactivates tuberous sclerosis complex 1/2 (TSC1/TSC2) [47] which stimulated Akt–mTOR signaling and autophagy in muscular dystrophy (Fig. 16.2), since signaling through IGF-1/PI3K/Akt activated the mechanistic target of rapamycin (mTOR) pathway [48] and critically mediated the fork head boxO (FoxO) transcription factors [49]. Protein synthesis in skeletal muscle is strongly related to Akt/PKB signaling, through the activation of mTOR [48], to regulate GSK3 (glycogen synthase kinase 3) activity which results in enhanced glycogen synthesis [50]. Furthermore, the defected of insulin actions may produce the increase of protein degradation [51]; this is supposed the Akt/FoxO pathway may contribute to sarcopenia-related muscle protein degradation.
16.3.3.2 FoxOs Transcription Factors
Decline of skeletal muscle mass or strength involves regulation of muscle protein catabolism and mitochondrial dysfunction. FoxOs are important regulators of muscle energy homeostasis and carbohydrate catabolism in the fast state [52]. FoxO1 and FoxO3 induced a decreasing of muscle mass which associated with an upregulation of ubiquitin E3 ligases MAFbx/atrogin-1 and the muscle ring-finger protein 1 (MuRF1) expression [53]. Recently, defect in autophagy-dependent signaling, an important mechanism for maintaining cell self-renew and protein turnover, is also observed in various muscular dystrophies [54]. The autophagic regulator proteins, LC3-II, Atg12, and Bnip3, promote mitochondrial disruption by activation of FoxOs during muscle atrophy [55]. Furthermore, mitochondrial dysfunction has been proposed through FoxOs, switching of muscle type from slow-twitch oxidative type I fiber to fast-twitch glycolytic fiber (MyHCI to MyHCII).
16.3.3.3 PGC-1α and AMPK
In sarcopenia and sarcopenic obese individuals, insulin resistance is involved in developing muscle atrophy in aging and diabetes mellitus [56]. Importantly, high-fat diet-induced insulin resistance observed the reduction of skeletal muscle mitochondrial function and decreased expression of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) [57]; thus, the hypothesis of mitochondrial dysfunction leading to sarcopenia has been in attention. The downregulation of PGC-1α by high-fat diet implicated the development of skeletal muscle insulin resistance of T2D individuals compared to nondiabetic individuals [58–61]. It was also suggested that PGC-1α plays an important role in fiber-type switching from fast-twitch glycolytic fiber into slow-twitch oxidative type I fiber [62]. These fiber-type switching effects through PGC-1α might be induced by PGC-1α1. However, muscle-specific induction of PGC-1α4, but not PGC-1α1, has shown the increase of muscle mass and strength, resistance to cancer cachexia through activating IGF-1, and suppressing myostatin gene expression [63]. Altogether, PGC-1α and its isoforms may play a crucial role in regulating mitochondrial biogenesis and muscle mass and strength.
AMP-activated protein kinase (AMPK) is a primary regulator of skeletal muscle metabolic homeostasis [64]. AMPK impacts the insulin-mediated effects on muscle protein synthesis through the interfering by the Akt/PKB signaling pathway [65] and promotes glucose uptake and oxidation through migration of GLUT4 to the cellular membrane [66]. Importantly, AMPK activation by AICAR in skeletal muscle [67, 68] improved muscle function and mitochondrial activity in muscle atrophy. In addition, AMPK inactivated mTOR and decreased the activation of ribosomal protein S6 kinase 1, an activator of protein synthesis, thereby increasing the rates of sarcopenic muscle mass loss. On the other hand, AMPK-mediated activation of FoxO3 contributed to the proteolysis with the expression of muscle atrophy F-box (MAFbx/atrogin-1) and MuRF1 [69]. AMPK also was found to promote directly the phosphorylation of PGC-1α and to induce mitochondrial biogenesis [70, 71]. Further investigations will be needed to clarify these conflicting findings on the role of AMPK in the development of sarcopenia.
16.3.4 ROS and Mitochondrial Dysfunction
ROS and oxidative stress have been considered as important pathogenic components of metabolic diseases [72]. Mitochondrial dysfunction produces increased amounts of ROS, resulting in oxidative damage, which includes decreased mitochondrial content and oxidative capacity and increased mitochondrial DNA mutations [73]. Activation of the renin-angiotensin system (RAS) is commonly observed in patients either with diabetes, obesity, or both. ROS production could be enhanced by angiotensin II (Ang II), which stimulated the angiotensin type 1 (AT1, G-protein-coupled) receptor and activated NADPH oxidase. In rodent model, RAS blockade increases survival rate and prevents age-related defects [74]. Thus, it is suggested that Ang II contributes to mitochondrial dysfunction in the aging process. This indicated that diabetes mellitus and age-related sarcopenia may have additive effects for ROS production. Furthermore, NF-kB induces muscle breakdown by promoting protein degradations in skeletal muscle through regulation of ubiquitin-proteasome pathway (UPS) [75]. The maintenance of mitochondrial morphology and regeneration of self-renewal could be due to the mitochondrial fusion and fission interactions. This mitochondrial fission contributes to the quality control of creating new mitochondria and removing of damaged mitochondria during high cellular stress [76]. Disruptions of either event may induce metabolic disorders and leads to developmental defects and diseases [77], suggesting that the maintenance in aging muscle cell mitochondrial morphology may prevent cell dysfunction.
16.3.5 AGE Accumulation and Diabetic Neuropathy
The formation of advanced glycation end products (AGEs) is generated through nonenzymatic glycation of many heterogenous compounds and the complex and diverse possibilities of reaction of glucose with proteins, lipids, and nucleic acids [78]. AGEs are regarded as key molecules to be one source of oxidative stress in aging [79]. It is well known that long-term high-fat diet and endogenously formed AGEs contribute to the progression of diabetic complications [80]. AGEs are produced endogenously via food, and the concentration of circulating AGEs increases in high-fat diet [81]. Interestingly, it has been reported that AGE accumulation was decreased in association with administration of insulin and greater in fast-twitch fibers in non-insulin diabetic animal model [82]. AGE/RAGE in aging stimulates the activation of ERK1/2 and P38 MAPK pathways and increased apoptosis transcription factor such as NF-kB [83]. In recent years, the modified protein methylglyoxal (MG) was observed to involve AGE formation in aged individuals [84, 85]. MG is believed to induce protein glycation leading to the formation of AGEs; some reports indicated that MG or AGEs administration to Sprague-Dawley rats resulted in increased glucose levels and insulin resistance [86, 87]. Moreover, the direct effects of AGEs from Tanaka’s group have shown that AGE2 or AGE3 markedly suppressed the expressions of MyoD and myogenin protein on myoblastic differentiation in C2C12 cells and significantly inhibited mRNA expression. This suggested that AGEs have direct negative effects on myogenesis [88].
T2D leads to chronic hyperglycemia and is related to the major age-related microvascular complications such as microangiopathic and macroangiopathic damage and motor neuropathies (diabetic neuropathy) [89]. Enhanced mitochondrial apoptosis has also been observed in muscle denervation and implicated in diabetes and neuropathy in human neuromuscular disorders, which exhibited significant muscle weakness and reduced functional capacity in the ankle and knee [90]. In addition, the AGE-RAGE axis and accumulation of AGEs in the peripheral nerve also play important roles in the pathogenesis of diabetic neuropathy [91], thus impairing the quality of life of diabetic patients. It is not yet established, but increasing studies suggested that accumulation of AGEs through glucose intolerance enhanced by mitochondrial ROS together with promotion of apoptosis leads to an elevated risk of developing sarcopenia.
16.4 Potential Treatments for Sarcopenia with Diabetes
Epidemiological and intervention studies for exercise training have strongly supported its efficacy for prevention, leading to the management of diabetes and sarcopenia. In exercising and amino acid supplementation (AAS) study [92], not only enhanced muscle mass or walking speed but also enhanced muscle strength was observed in sarcopenic women. In addition, high-intensity progressive resistance training was effective in improving glycemic control and physical activity, increasing lean mass among high-risk older adults with T2D [93]. Thus, resistance training and a combination of training and nutritional replenishment, like amino acids, might also be a beneficial intervention in sarcopenia with diabetes.
As for antidiabetic drugs, a class of thiazolidinediones (TZDs) can activate AMPK in insulin-resistant animals [94] and mediate mitochondrial effects on neurodegeneration [95]. In T2D patients, rosiglitazone [96] and pioglitazone [97] both enhanced insulin reactions and reduced plasma nonesterified fatty acids [97], which improved insulin-stimulated muscle glucose disposal. These TZDs may involve a potential role of age-related mitochondrial dysfunction, neurodegeneration diseases [98], and diabetes with sarcopenia. Indeed, a combination of pioglitazone and resistance training leads to a potentiated effect on muscle power compared with resistance training alone in older obese women [99]. Metformin also activates AMPK and enhances insulin sensitivity in skeletal muscle, thereby stimulating glucose uptake [100]; however, there has been no reliable clinical evidence so far. At any rate, insulin resistance and glucose intolerance can contribute to muscle wasting; therefore, the therapies for improving insulin resistance and glucose intolerance are supposed to have a potential to prevent sarcopenia.
16.5 Closing Remarks and Perspectives
We conclude that subjects with sarcopenia in diabetes mellitus would have higher physical dysfunction and mortality risks than those nondiabetic older adults. Sarcopenic obesity could be described as independent of sarcopenia and obesity, both related to insulin resistance and inflammation in diabetes mellitus. We introduce the molecular pathways underlying the pathogenesis of sarcopenia in diabetes in this chapter and, in particular, mitochondrial dysfunction and AGE accumulation might be significant targets common to sarcopenia and diabetes. Further studies will be needed to improve our knowledge on the interaction between diabetes and sarcopenia and to establish beneficial therapeutic interventions combined with exercise for slowing down and reversing the loss of muscle mass and strength in older adults with T2D.
References
1.
United Nations (2007) World population prospects, the 2006 revision: highlights. United Nations, New York
2.
Smalley KJ, Knerr AN, Kendrick ZV, Colliver JA, Owen OE (1990) Reassessment of body-mass indexes. Am J Clin Nutr 52(3):405–408
3.
Dutta C, Hadley EC, Lexell J (1997) Sarcopenia and physical performance in old age: overview. Muscle Nerve 5:S5–S910.1097/01.mco.0000109601.04238.46 CrossRef
5.
Berger MJ, Doherty TJ (2010) Sarcopenia: prevalence, mechanisms, and functional consequences. Interdiscip Top Gerontol 37:94–114. doi:10.1159/000319997 CrossRef
6.
Villareal DT, Banks M, Siener C, Sinacore DR, Klein S (2004) Physical frailty and body composition in obese elderly men and women. Obes Res 12(6):913–920. doi:10.1038/oby.2004.111 CrossRef
7.
Goodman MN, Ruderman NB (1979) Insulin sensitivity of rat skeletal muscle: effects of starvation and aging. Am J Physiol 236(5):E519–E523
8.
Sial S, Coggan AR, Carroll R, Goodwin J, Klein S (1996) Fat and carbohydrate metabolism during exercise in elderly and young subjects. Am J Physiol-Endocrinol Metab 271(6):E983–E989
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:
 Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
 Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
 Overview
Overview
 Fracture Risk in Diabetes
Fracture Risk in Diabetes
 Body Temperature Regulation During Exercise Training
Body Temperature Regulation During Exercise Training
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


