Rheumatic Diseases of Childhood
James T. Cassidy
The rheumatic diseases of childhood comprise a group of heterogeneous disorders that result in inflammation of the connective tissues of the body. A frequent manifestation of these diseases is arthritis: the objective inflammation of a joint. Although the etiologies of these disorders are unknown, many have an immunogenetic predisposition and are characterized by prominent autoimmune phenomena: circulating autoantibodies such as antinuclear antibodies (ANAs) and rheumatoid factors (RFs) and, in some cases, the deposition of immunoglobulins in affected tissues. Often, tissue accumulation of lymphocytes and plasma cells occurs. Usually, these rheumatic diseases are chronic and seemingly self-perpetuating, although many respond to nonsteroidal antiinflammatory drugs (NSAIDs), glucocorticoids, antiinflammatory drugs, or immunosuppressive agents.
Data on the prevalence of the rheumatic diseases in children are incomplete. Juvenile rheumatoid arthritis (JRA) is the most common disorder for which reasonable estimates of frequency have been published. A study from Minnesota cited an incidence of 13.9 per 100,000 children per year (95% confidence limits: 9.9 to 18.8) and a prevalence of 113.4 per 100,000 children (95% confidence limits: 69.1 to 196.3). A Finnish investigation estimated its incidence to be 18.2 per 100,000 children per year, and the incidence of all forms of arthritis in childhood to be 108.5 per 100,000.
The prevalence of the other rheumatic diseases can be estimated by noting their relative frequency of referral to pediatric rheumatology clinics. Table 434.1 includes data from children referred to these clinics in North America from 1992 to 2002.
Table 434.2 is a condensed diagnostic classification of juvenile arthritis. Each of the major types of rheumatic diseases
is included, but only selected subtypes pertinent to pediatric practice are provided for each category. As in all other areas of medicine, the correct diagnosis of a disease without a pathognomonic finding or known etiology requires the careful exclusion of similar disorders. Most of the rheumatic diseases occur more commonly in girls than in boys. However, exceptions occur, such as for polyarteritis and ankylosing spondylitis.
is included, but only selected subtypes pertinent to pediatric practice are provided for each category. As in all other areas of medicine, the correct diagnosis of a disease without a pathognomonic finding or known etiology requires the careful exclusion of similar disorders. Most of the rheumatic diseases occur more commonly in girls than in boys. However, exceptions occur, such as for polyarteritis and ankylosing spondylitis.
TABLE 434.1. FREQUENCY OF THE PEDIATRIC RHEUMATIC DISEASES | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||
TABLE 434.2. DIAGNOSTIC CLASSIFICATION OF JUVENILE ARTHRITIS | |
|---|---|
|
In JRA, the primary focus of the clinical disease is on arthritis: synovitis of the joints of the appendicular skeleton. Arthritis must be distinguished from arthralgia, which is pain in a joint without objective findings of inflammation on physical examination. Many more children have transient episodes of arthralgia, or even of arthritis, than can be categorized as JRA. One study estimated that only approximately 15% of children with unexplained arthralgia go on to develop chronic arthritis that meets the classification criteria for JRA.
Although arthritis is a frequent characteristic of the other rheumatic diseases, often they have more prominent manifestations of inflammation in other organ systems (Table 434.3). In systemic lupus erythematosus (SLE), the primary focus is immune-complex vasculitis of the arterioles that underlie the cutaneous, mucosal, and serosal surfaces of the body and involvement of internal organs, such as the kidneys and central nervous system (CNS). In dermatomyositis, the affected organs primarily are the skeletal muscles, skin, and gastrointestinal tract. In scleroderma, the skin, gastrointestinal tract, and cardiopulmonary system primarily are involved. In idiopathic or primary vasculitis, blood vessels are the predominant target organs, with multisystem disease involving the CNS; cardiopulmonary, gastrointestinal, and renal systems; and the skin, depending on the specific type of vasculitis in the child.
TABLE 434.3. CLINICAL MANIFESTATIONS OF CONNECTIVE TISSUE DISEASES | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
JUVENILE RHEUMATOID ARTHRITIS
JRA is the most common pediatric rheumatic disease having arthritis as the principal manifestation. It is one of the more frequent chronic childhood illnesses and a leading cause of disability and blindness.
Pathogenesis
The etiology of JRA is unknown. It likely does not represent a single disorder but rather a spectrum of diseases of diverse pathogenesis. An immunogenetic predisposition is present in certain children: the HLA antigens DR5 (DRB1*1104), DRw6, DRw8, DQw1 (DQA1*0501), and DPw2.1 (DPB1* 0201) are associated with the development of persistent oligoarthritis at an early age in young girls with ANA seropositivity, and DR4 (DRB1*0401 and 0404) is increased in frequency in older children with polyarthritis who are seropositive for RFs. The risk associated with these immunogenetic profiles appears to be age-related. A JRA-like arthritis is a manifestation of
immunodeficiency in children with selective IgA deficiency, agammaglobulinemia or hypogammaglobulinemia, and C2 complement component deficiency. In children with oligoarthritis, T-lymphocyte proliferative responses to heat-shock protein 60 are associated with a remission of the inflammatory disease. A study of 144 families has established the association of several tumor necrosis factor-alpha (TNFα) single nucleotide polymorphisms (SNPs) with juvenile oligoarthritis. Interleukin-6 (IL-6) is increased in the circulation of children with systemic-onset disease. In systemic-onset disease, IL-6-174G is over-represented in the transcriptional regulation of the IL-6 gene and is associated with resistance to glucocorticoid therapy. Also in systemic-onset disease, high levels of macrophage inhibitory factor (MIF) in serum and synovial fluid are associated with a G to C SNP at position-173 of the MIF gene that leads to glycocorticoid resistance, persistence of arthritis, and a relatively poor outcome.
immunodeficiency in children with selective IgA deficiency, agammaglobulinemia or hypogammaglobulinemia, and C2 complement component deficiency. In children with oligoarthritis, T-lymphocyte proliferative responses to heat-shock protein 60 are associated with a remission of the inflammatory disease. A study of 144 families has established the association of several tumor necrosis factor-alpha (TNFα) single nucleotide polymorphisms (SNPs) with juvenile oligoarthritis. Interleukin-6 (IL-6) is increased in the circulation of children with systemic-onset disease. In systemic-onset disease, IL-6-174G is over-represented in the transcriptional regulation of the IL-6 gene and is associated with resistance to glucocorticoid therapy. Also in systemic-onset disease, high levels of macrophage inhibitory factor (MIF) in serum and synovial fluid are associated with a G to C SNP at position-173 of the MIF gene that leads to glycocorticoid resistance, persistence of arthritis, and a relatively poor outcome.
Pathology
The synovial membrane in JRA is characterized by villous hypertrophy and hyperplasia of the synovial lining layer. Edema and hyperemia are present, along with vascular endothelial hyperplasia and the infiltration of lymphocytes and plasma cells. Pannus formation eventually occurs in severely affected joints and leads to the destruction of articular cartilage and contiguous bone.
Infiltrates of inflammatory cells occur also in parenchymal organs, such as the liver, and along serosal surfaces of the pericardium, pleura, and peritoneum. Rheumatoid nodules result from a small-blood-vessel vasculitis but are rare findings in affected children. The rash of JRA is characterized histologically by a neutrophilic perivasculitis and infiltration of round cells surrounding capillaries and subdermal venules.
Clinical Manifestations and Complications
Although the onset of JRA occurring before a child reaches 6 months of age is unusual, the mean age at onset characteristically is young (1 to 3 years), with a substantial number of cases beginning throughout childhood and young adolescence. Girls are affected at least twice as frequently as are boys.
Fatigue, low-grade fever, anorexia, weight loss, and failure to grow are common findings at onset of the disease in moderate to severely affected children. Morning stiffness, musculoskeletal gelling after inactivity, and night pain often are associated with incompletely controlled disease. Affected children may not, however, always communicate these symptoms to their parents. Instead, the child may present with increased irritability, a posture of guarding the joints, a limp, or refusal to walk.
TABLE 434.4. CLASSIFICATION OF TYPES OF ONSETS OF JUVENILE RHEUMATOID ARTHRITIS | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
Tenosynovitis also is a component of active disease. A stenosing synovitis of the flexor tendon sheaths may lead to loss of extension of the fingers or to a trigger finger. Rheumatoid nodules may occur on the tendons or subcutaneously over pressure points. They are found particularly in children who have widespread polyarthritis, are older at onset, and have prominent small-joint disease, an unrelenting course with early development of bony erosions, and RF seropositivity.
Types of Onset
The classification of JRA is based on recognition of at least three distinct types of onsets of disease (Table 434.4): polyarthritis, oligoarthritis (pauciarticular disease), and systemic disease. These types of onsets are characterized by specific signs and symptoms at presentation and during the first 6 months of illness.
Polyarthritis is disease that begins in five or more joints. This onset occurs in one-third of affected children and may be acute or insidious. Systemic manifestations are not severe or persistent. The arthritis generally involves large joints, such as the knees, wrists, elbows, and ankles, and usually is symmetric. However, the smaller joints of the hands or feet may be affected early or late. Younger children may not complain of pain, although the joints are tender and appear painful on motion. The cervical spine often is involved in this type of onset, although onset of JRA solely in the cervical spine is a rare occurrence. The neck may be painful or stiff, with an alarmingly rapid loss of extension and rotation. Atlantoaxial subluxation may occur early and places a child at risk for incurring an injury of the cervical cord in an accident or with attempted intubation before general anesthesia. Temporomandibular joint disease is a relatively common occurrence in children with polyarthritis and leads to limitation or asymmetry of bite and micrognathia.
The onset of JRA in one-half of affected children involves four or fewer joints. Oligoarthritis or pauciarticular disease often is confined to the knees or ankles, or it may involve a single joint, such as a knee, at onset and throughout the course. The hips usually are spared at onset. Extra-articular systemic disease, except for chronic uveitis, is a distinctly unusual finding unless the child goes on to develop polyarticular involvement (approximately 10%).
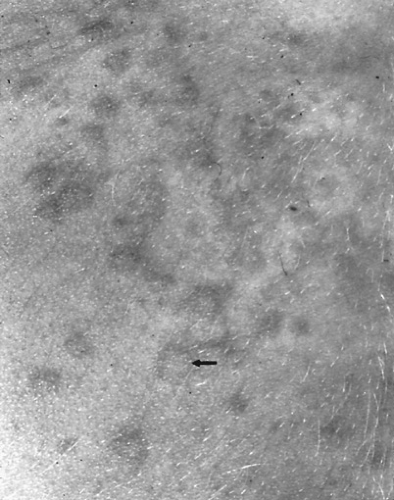 FIGURE 434.1. Rash of systemic-onset JRA. This 4-year-old girl presented with high-spiking fever that occurred once a day, accompanied by a transient nonpruritic rash. The arrow points to central clearing in a lesion. (Reprinted with permission from Cassidy JT. Juvenilerheumatoid arthritis. In Ruddy S, Harris ED Jr., Sledge CB, eds. Kelley’s textbook of rheumatology. Philadelphia: WB Saunders, 2001.) |
A small number of children have onset of JRA with severe constitutional and systemic disease. These systemic signs and symptoms may precede the appearance of overt arthritis by weeks, months, or even years. A hallmark of this type of disease is a high-spiking fever, often combined with a rheumatoid rash. Elevations in temperature occur once or twice a day, often in late afternoon or evening, to a level of 39°C or higher, with a quick return to baseline temperature or lower. This quotidian pattern is highly suggestive of a diagnosis of JRA.
The rash of JRA develops often with this fever and consists of 2- to 5-mm erythematous morbilliform macules (Fig. 434.1). It is seen most commonly on the trunk and proximal extremities, and over pressure areas, but it may occur on the face, palms, or soles. Generally, it is not pruritic. The most characteristic feature is its transient nature. Usually, any single lesion does not persist for longer than an hour. Sometimes, the rash can be elicited in a child by rubbing or scratching the skin: the isomorphic response or Koebner phenomenon. This rash never occurs in children with oligoarthritis.
Children with systemic onset usually have concurrent hepatosplenomegaly and lymphadenopathy. Pericarditis, hepatitis, and other visceral disease may develop. Pulmonary involvement consists of a wide spectrum of abnormalities, such as pleuritis and effusion, interstitial fibrosis, and hemosiderosis. The CNS may be affected, but distinguishing encephalopathy from drug toxicity, viral infection, or other complications of the systemic illness and fever may be difficult.
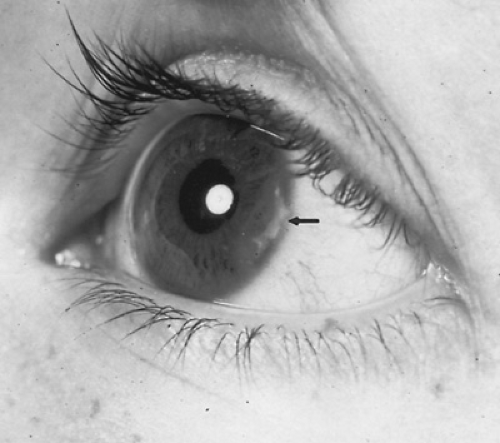 FIGURE 434.2. The arrow points to an area of band keratopathy just inside the corneal limbus in a girl who had ANA-positive oligoarticular JRA. Her chronic uveitis was bilateral and had resulted in a decrease in vision to 20/400 in the right eye. |
Complications
Chronic Uveitis
One of the most serious complications of JRA is the development of a chronic nongranulomatous uveitis involving the iris, ciliary body, and occasionally the posterior choroid (Fig. 434.2). Involvement usually is bilateral and can lead to blindness. Chronic uveitis characteristically has an insidious, asymptomatic onset and is diagnosed only by ophthalmologic and slit-lamp examinations at the time of diagnosis; these examinations must be repeated at frequent intervals during the first years after onset. Chronic uveitis is confined to children with polyarthritis or oligoarthritis. It tends to occur particularly in young girls with an early age of onset, limited joint disease, and ANA seropositivity.
Growth Retardation
Disturbances of growth and normal development are complications of any chronic disease and often are prominent in children with JRA. Linear growth is retarded during periods of active disease or with the use of glucocorticoid drugs. Localized growth retardation also may occur in specific areas such as the jaw (micrognathia). Unequal leg or arm lengths develop with monarticular disease. Often, the development of secondary sexual characteristics is delayed. Psychosocial retardation may be a frequent, persistent, and potentially severe complication. Psychological regression to a more infantile pattern is present in most children with moderate to severe disease. Osteopenia is an almost universal occurrence and, if persistent, is a risk factor later in life for fractures and premature osteoporosis. Children with chronic arthritis have elevated levels of osteoprotegerin, a decoy receptor, that inhibits the differentiation and activation of osteoclasts. This abnormality is related to a TC that correlates with low bone mass and bone erosions.
Laboratory Examination
Children with active disease develop a normocytic, hypochromic anemia characteristic of the chronic anemia of inflammation. Often, it is moderately severe, with the hemoglobin concentration in the range of 7 to 10 g/dL. Leukocytosis and
thrombocytosis are common findings with active disease, but they are much less pronounced in children with oligoarthritis.
thrombocytosis are common findings with active disease, but they are much less pronounced in children with oligoarthritis.
The acute-phase reactants often are elevated at the onset of disease and are moderately useful in following the course of the inflammation. The Westergren erythrocyte sedimentation rate (ESR), C-reactive protein level, and immunoglobulin concentrations reflect inflammatory activity if increased. Serum complement components usually are elevated at onset and with exacerbations. The serum amyloid-like protein (SAA) is increased in concentration in children with active disease. Soluble immune complexes can be detected in the sera of some children, particularly in those with systemic onset.
Tests for RFs are positive in children with JRA less frequently than in adults with rheumatoid arthritis, although approximately 10% of children eventually become seropositive. RFs are IgM macroglobulins with antigenic specificity directed against the unfolded H-chains of IgG. Seropositivity seldom is observed in a child younger than 7 years of age; RFs tend to be present in children who are older at onset and in those who have symmetric polyarthritis with involvement of the small joints, subcutaneous rheumatoid nodules, articular erosions, and a poor functional outcome.
ANA seropositivity is present in at least 45% of children with JRA. The pattern of fluorescent staining usually is homogeneous or speckled; the titer generally is low to moderate. The presence of these antibodies is a significant risk factor for the development of chronic uveitis. They are found less commonly in older boys and in children with systemic disease. A positive ANA determination is a valuable diagnostic measure in a child suspected of having JRA because this antibody activity is not found frequently in other childhood illnesses, except for the rheumatic diseases and transient acute viral infection.
The synovial fluid white cell count in JRA is elevated moderately, in the range of 10,000 to 20,000/mm3. Synovial glucose concentration is low. Complement levels may be depressed, as evidence for intrasynovial complement activation.
Urinalysis generally is normal in children with JRA except for those few who have a mild glomerulitis at onset. Proteinuria also may occur with fever. Persistent proteinuria may be the first evidence of drug toxicity or amyloidosis. Renal papillary necrosis may develop related to NSAID therapy and dehydration during the course of the disease.
Radiologic Examination
Children with JRA exhibit a wide range of distinctive radiologic findings. Early changes consist of soft-tissue swelling, juxta-articular osteoporosis, and periosteal new-bone apposition. Development of the ossification centers may be accelerated, or premature epiphyseal closure may be present, leading to stunting of bone growth. In children with polyarthritis or systemic disease, especially later in the course, marginal erosions may develop, along with narrowing of the cartilaginous spaces.
Cervical spine disease is characteristic of JRA. The upper cervical segments principally are affected with apophyseal joint fusion and atlantoaxial subluxation (Fig. 434.3). The lower vertebrae also may be involved, with failure to grow normally. Sacroiliac arthritis in children with JRA is not characterized by the degree of involvement and reactive sclerosis that would be seen in ankylosing spondylitis. Fractures, particularly of the long bones and vertebrae, occur in children who develop generalized osteopenia.
Routine radiographic studies generally are sufficient to document this progression of abnormalities in evaluating a child’s response to a management program. Newer methods of objectively evaluating joint disease (e.g., bone scans, computed tomography [CT], and magnetic resonance imaging [MRI]) can be useful diagnostically. MRI is more precise than is routine radiographic examination in delineating soft-tissue abnormalities in response to abnormal bone growth or fusion and in detecting early cartilage or bone destruction.
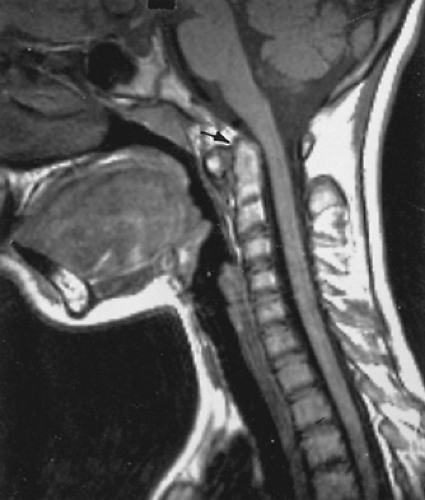 FIGURE 434.3. Magnetic resonance image of the cervical spine of a child with 7 mm of atlantoaxial subluxation. The arrow points to the odontoid, which is beginning to impinge on the upper cervical cord. |
Diagnosis
The classification criteria for JRA of the American College of Rheumatology (ACR) and for juvenile idiopathic arthritis of the International League for Arthritis and Rheumatism (ILAR) are compared in Table 434.5. JRA was defined by the ACR as onset of arthritis in a child younger than 16 years. Arthritis is defined clinically by swelling or effusion or the presence of two or more of the following signs: limitation of range of motion, tenderness or pain on motion, and increased heat in one or more joints. JRA often is a diagnosis of exclusion and, therefore, similar diseases must be considered (see Table 434.2). The differential diagnosis generally includes other rheumatic
and connective tissue diseases, especially acute rheumatic fever, SLE, and ankylosing spondylitis. In our experience, other forms of arthropathy, such as reactive and psoriatic arthritis, are uncommon findings. Particular attention must be accorded, however, to infectious arthritis, serum sickness, Henoch-Schönlein purpura, the enteropathies such as ulcerative colitis and regional enteritis, and hematologic diseases such as leukemia, sickle-cell anemia, and hemophilia. The concurrence of arthritis and immunodeficiency already has been mentioned. Certain viral illnesses, especially rubella, mumps, and Lyme disease, have an associated arthritis. Tumors, especially neuroblastoma in young children, may present as bone pain or arthritis. Although the onset of JRA in the hip is an uncommon occurrence, transient synovitis, Legg-Calvé-Perthes disease, and slipped capital femoral epiphysis may mimic JRA, especially if the pain is referred to the knee, as it often is.
and connective tissue diseases, especially acute rheumatic fever, SLE, and ankylosing spondylitis. In our experience, other forms of arthropathy, such as reactive and psoriatic arthritis, are uncommon findings. Particular attention must be accorded, however, to infectious arthritis, serum sickness, Henoch-Schönlein purpura, the enteropathies such as ulcerative colitis and regional enteritis, and hematologic diseases such as leukemia, sickle-cell anemia, and hemophilia. The concurrence of arthritis and immunodeficiency already has been mentioned. Certain viral illnesses, especially rubella, mumps, and Lyme disease, have an associated arthritis. Tumors, especially neuroblastoma in young children, may present as bone pain or arthritis. Although the onset of JRA in the hip is an uncommon occurrence, transient synovitis, Legg-Calvé-Perthes disease, and slipped capital femoral epiphysis may mimic JRA, especially if the pain is referred to the knee, as it often is.
TABLE 434.5. COMPARISON OF ACR AND ILAR CRITERIA FOR ARTHRITIS IN CHILDREN | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
Therapy
Conservative management attempts to control the clinical manifestations of the disease and prevent or minimize deformity. Ideally, this approach involves a multidisciplinary team that follows the affected child throughout the course of the illness. Management should be family-centered, community-based, and coordinated. The long-term therapeutic program must be accepted by the child and family and judged to have a favorable risk-benefit ratio by the pediatric rheumatologist.
Prognosis is reasonably satisfactory for most children with JRA, so the philosophy of management initially should stress the simplest, safest, and most conservative measures. If this program of treatment proves inadequate, other therapeutic modalities should be chosen (Table 434.6).
NSAIDs are moderately effective in suppressing inflammation and fever. Aspirin seldom is used in North America at this time, and drugs such as naproxen, ibuprofen, and tolmetin are preferred. The first two drugs can be prescribed also as a suspension, which is particularly useful for younger children. All children with JRA should receive yearly influenza vaccines and should be immune to varicella.
Hydroxychloroquine is a useful adjunctive agent for treating the child with more progressive disease. Because of concern over retinopathy as a toxic reaction, a relatively low dose is chosen, (e.g., 5 mg/kg/day), and every 6-month ophthalmologic examinations, especially for color vision, are monitored during its administration. Intramuscular gold compounds and D-penicillamine were used previously for children whose polyarthritis was unresponsive to conservative management. Toxicities from these drugs primarily are hematologic, renal, or hepatic.
TABLE 434.6. MANAGEMENT OF CHILDREN WITH JUVENILE RHEUMATOID ARTHRITIS | |
|---|---|
|
Methotrexate has become the most accepted approach to the advanced drug treatment of severe or resistant polyarthritis. In a large, multinational, double-blind, randomized trial, a dose of 10 mg/m2 once a week markedly reduced the articular severity index, compared with placebo. This drug is attractive for pediatric use because it is given only once a week orally as a pill or liquid, and it has no proven oncogenic potential or untoward risk of causing sterility. Toxicity is monitored clinically and by periodic complete blood cell counts and liver enzyme determinations.
Additional therapy can include etanercept, a TNFα blocker. It consists of two TNFα receptor monomer chains fused to the Fc domain of human IgG1. The drug binds TNFα in the vascular and extracellular compartments and inhibits its activity. A controlled trial of etanercept in children with polyarticular JRA who had failed therapy with methotrexate indicated that 74% demonstrated improvement at 3 months on a subcutaneous dose of 0.4 mg/kg twice a week. A 2-year follow-up indicated that the overall rate of improvement was 79% and remissions occurred in 37%.
Infliximab is a monoclonal antibody that has the capacity to bind TNFα in both the extracellular and vascular compartments, and also on the cell surface. A pediatric trial is currently in progress to evaluate the safety and efficacy of this drug in JRA. Another biologic response modifier is anakinra (r-metHuIL-1ra), which interferes with the actions of IL-1 in the inflammatory response.
The long-term safety of cytokine blockade is unknown. Potentially, it is associated with an increased frequency of systemic infection and should not be used in children with recurrent or chronic infections. Tuberculosis always should be excluded before beginning therapy.
Glucocorticoid drugs should be reserved for treatment of the severely involved child who is recalcitrant to more conservative therapeutic regimens or for those with life-threatening disease and its complications, such as pericarditis. Many toxicities (e.g., Cushing syndrome) and growth retardation are associated with their use. Ophthalmic administration is indicated for treatment of chronic uveitis. Intra-articular steroid often is used to achieve the specific goals of a physical therapy program or for persistent monarticular or oligoarticular involvement.
Other immunosuppressive agents seldom are required in the long-term treatment of JRA. However, some children have failed to improve on all other therapies and are candidates for experimental protocols involving agents such as azathioprine or cyclophosphamide. Critical considerations are the oncogenic potential of these agents, potential sterility, and bone marrow suppression. Intravenous immunoglobulin, cyclosporin A, and other biologic response modifiers are additional approaches to experimental therapy.
Physical and Occupational Therapy
The maintenance of function and prevention of deformity cannot be overemphasized in the total management of the child with JRA. Appropriate prescriptions for physical and occupational therapy, a balanced program of rest and activity, and selective splinting are indicated. Normal play should be encouraged. Only unacceptable levels of stress on inflamed weight-bearing joints should be limited. Children with cervical spine disease should wear a padded collar when traveling in an automobile or studying.
Reconstructive Surgery
Synovectomy or tenosynovectomy sometimes is indicated in the course of management. Total joint replacement generally is delayed until after growth has ceased and the epiphyses have closed. Cosmetic surgery and orthodontic reconstruction for micrognathia or destruction of the temporomandibular joint are useful late approaches when indicated.
Prognosis
In general, more than half of the children who develop JRA have a satisfactory recovery from their disease and enter adult life without serious functional disability, although as many as 45% may have continuing low-grade activity for some years. A small percentage of patients whose disease has gone into remission will have a recurrence of arthritis during the adult years. As many as 15% of children with JRA, however, enter adulthood with significant functional disability. The child most at risk has had polyarthritis of later age of onset, early symmetric involvement of the small joints of the hands or feet, unremitting activity of the joint disease, early appearance of erosions, prominent systemic manifestations, and development of RF seropositivity and subcutaneous nodules. Progressive hip disease also is a major cause of long-term disability.
Serious functional disability is an uncommon development in children who have oligoarthritis and pursue a course of limited joint disease. The prognosis for sight in children with chronic uveitis has improved dramatically, a development probably related to earlier detection and better management of this complication. Blindness, however, still occurs in as many as 15% of affected eyes.
Approximately one-half of children with systemic onset eventually will recover completely; however, most JRA-related deaths in the United States have been associated with this type of disease. Death occurs in fewer than 1% of children with JRA, and it often is a result of overwhelming infection. In Europe, renal failure secondary to amyloidosis is a leading cause of death. Amyloidosis seldom occurs as a complication of JRA in North America.
Estimates of prognosis should remain optimistic, and, in many children, prognosis is excellent. Of importance is that both the child and parents understand the disease and share in its long-term management. The nature of JRA, the goals of therapy, and the expected course of the disease should be discussed in detail. A carefully selected program of coordinated care, initiated by the pediatric rheumatologist, must be reinforced constantly by the team nurse and social worker. Some families of children with JRA display psychiatrically important disruptions (e.g., divorce, separation, and depression). Thus, a priority in the management of a child with this chronic illness is to foster normal psychological and social development and peer group activities. A child’s unceasing potential for physical and psychological growth is a critical factor working in favor of the therapeutic program and team management. This natural endowment for future physical and psychological development is what enables so much to be accomplished in most children with JRA.
SYSTEMIC LUPUS ERYTHEMATOSUS
SLE is a multisystemic disease in which widespread inflammatory involvement of the connective tissues and immune-complex vasculitis occur. It is a prototypic example of autoimmunity in humans that results from abnormal immunologic hyperreactivity and an immunogenetic predisposition to the disease.
Pathogenesis
Numerous environmental, hereditary, and immunogenetic factors are implicated in the pathogenesis of this autoimmune disorder. The F1 hybrid of New Zealand white and black mice develops an SLE-like disease with early onset of renal failure that is more marked in females than in males. Household dogs also develop an SLE-like illness and, identical to their mouse counterparts, have circulating antibodies to native DNA and immune-complex deposition in tissues.
Environmental triggers, such as excessive exposure to the sun, a drug reaction, or an infection, may precipitate the onset of SLE. The basic pathogenesis of SLE appears to be immunologic abnormalities of homeostatic control affecting nuclear and cytoplastic antigens. Antibodies found in children with SLE include those that are tissue-specific as well as those related to nuclear antigens. These antibodies participate in specific manifestations of the disease, such as acute hemolytic anemia, thrombocytopenia, leukopenia, and thrombosis, and in the antigen–antibody complex deposition that results in systemic vasculitis and lupus nephritis, as well as impaired cell-mediated immunity and T-suppressor cell dysfunction.
Another connective tissue disease or an abnormal marker of immunologic function (e.g., ANA) is present in approximately 10% of family members. SLE has occurred in identical twins (concordance 24%). It is associated with sporadic or familial immunodeficiency, such as selective IgA deficiency, and inherited disorders of complement components, such as homozygous C2, C1q, C1r, or C1 esterase inhibitor deficiency. In many affected children, a marked immunogenetic predisposition is evident in the extended haplotype for genes located on chromosome 6: HLA-B8, DR2 or DR3, DQw1 or DQw2, and C4A null.
Pathology
An immune-complex vasculitis with fibrinoid necrosis is the basic inflammatory lesion. Vascular lesions are widespread throughout the parenchymal organs, in subdermal tissues, and on the mucosal and serosal surfaces. Soluble immune complexes can be demonstrated beneath the vascular endothelium and along the dermal-epidermal junction in the lupus-band test.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


