Retinoblastoma
Murali M. Chintagumpala
EPIDEMIOLOGY
Retinoblastoma is a rare, highly malignant tumor of the retina of young children. Retinoblastoma is the seventh most common pediatric malignancy in the United States. The worldwide incidence of retinoblastoma is relatively stable at 1 case per 18,000 to 30,000 live births. No significant difference in incidence exists between sexes or among races. The average age at presentation ranges between 13 and 18 months; more than 90% of the cases are diagnosed before age 5 years. Retinoblastoma is a relatively slow-growing tumor that usually remains confined to the eye for months or even years. With early diagnosis, the overall 5-year survival rate exceeds 90%.
Retinoblastoma occurs unilaterally in 70% to 75% of cases. In more than 70% of cases, the tumor originates from a single focus and, when clinically detected, involves more than one-half of the retina, with extension into the vitreous chamber. Multifocal involvement may be observed with unilateral retinoblastoma, but is more common with bilateral disease. Although multiple tumor foci usually present simultaneously, in as many as 25% of cases, new foci may develop within weeks to months after the original diagnosis. The potential for metachronous occurrence of retinoblastoma warrants careful follow-up examination, even of the previously unaffected retina. Bilateral disease is detected at an earlier age (median, 13 months) than unilateral disease (median, 24 months). The long-term outlook for bilateral disease is significantly worse than for unilateral disease because of an increased incidence of second, nonocular malignant tumors in bilaterally affected cases.
Genetics
Retinoblastoma occurs in both hereditary (germinal) and nonhereditary (nongerminal) forms. Knudson has postulated a “two-hit” mutational event as being necessary for the development of disease. The retinoblastoma gene locus resides on human chromosome 13 at band q14. This retinoblastoma gene has been further characterized, mapped, and cloned and is the prototype for a class of recessive human cancer genes (tumor suppressor genes) in which a loss of activity of both normal alleles is thought to be associated with tumor genesis. Retinoblastoma gene mutations also have been found in some osteosarcomas, soft-tissue sarcomas, breast carcinomas, small cell carcinomas of the lung, and prostatic carcinomas.
Between 85% and 90% of the patients have no family history of retinoblastoma and represent the first mutational event within the family. All patients with bilateral retinoblastoma (25% of all patients with retinoblastoma) and approximately 15% of the patients with unilateral retinoblastoma harbor a germinal mutation. According to Knudson’s hypothesis, a somatic mutation after the germinal mutation is necessary for malignant transformation to occur. Approximately 85% of sporadic unilateral cases are nongerminal; according to Knudson’s hypothesis, two somatic mutations are required to produce the disease in these patients. When patients with either bilateral or unilateral retinoblastoma have the germinal mutation, 50% of their offspring will be affected by the disease, and 1 in 100 will harbor a gene but not express the disease. Direct analysis of the genetic defect in the genomic DNA is available to predict familial predisposition to retinoblastoma.
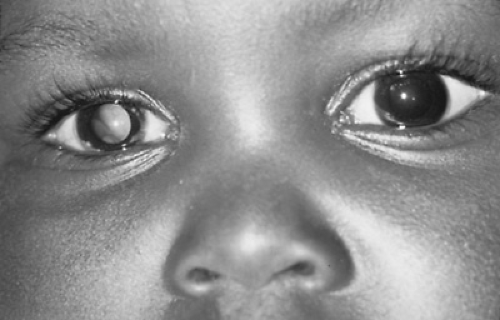 FIGURE 310.1. Abnormal white reflex, leukokoria, in a 7-month-old child with unilateral retinoblastoma. |
Perhaps 5% of patients with retinoblastoma present with additional abnormalities, including developmental delay, abnormal dermatoglyphics, imperforate anus, and failure to thrive. Constitutional deletions of the long arm of chromosome 13 have been found in these patients.
CLINICAL MANIFESTATIONS AND COMPLICATIONS
In the United States, most children with retinoblastoma are first identified by their parents. The most common presenting sign is a white pupillary reflex called leukokoria (Fig. 310.1 and Box 310.1). This abnormal reflex, present in 60% of patients, is the result of a centrally located tumor at the posterior pole. Replacement of the vitreous with tumor or retinal detachment also may be noted.
The second most common sign is strabismus, present in 20% of patients. In children younger than 4 years, strabismus is usually the result of esotropia. With retinoblastoma, both esotropia and exotropia may occur and usually indicate tumor involvement of the macular area. Other signs include a red, painful eye with glaucoma (7%), poor vision (5%), unilateral dilated pupil, heterochromia (different-colored irises), or nystagmus. Children with advanced stages of disease may present with signs of lethargy, anorexia, failure to thrive, neurologic defects, orbital mass, proptosis, or blindness.
BOX 310.1 Presenting Signs and Symptoms of Retinoblastoma
| ||||||||||||||
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


